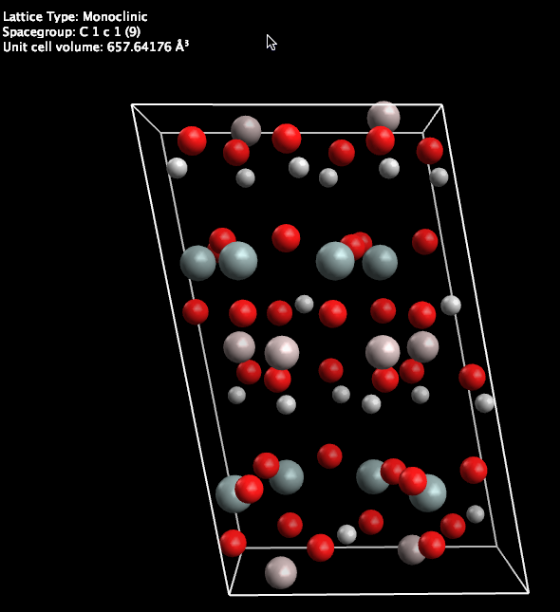
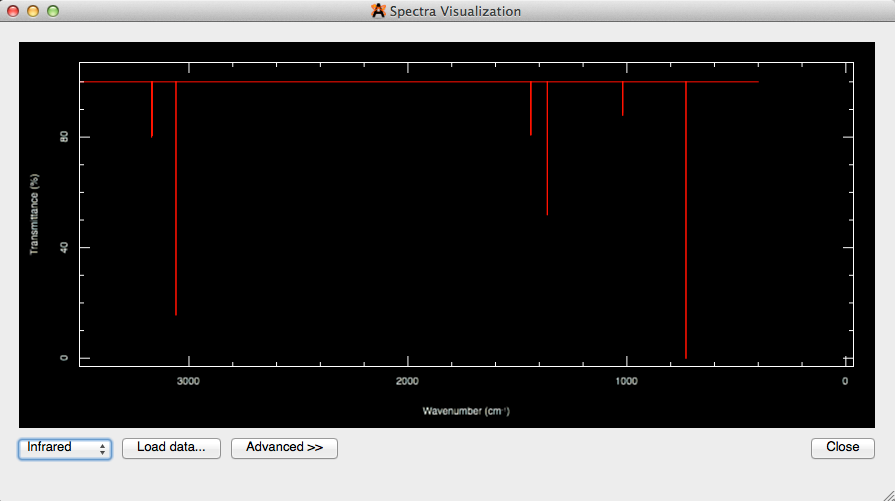
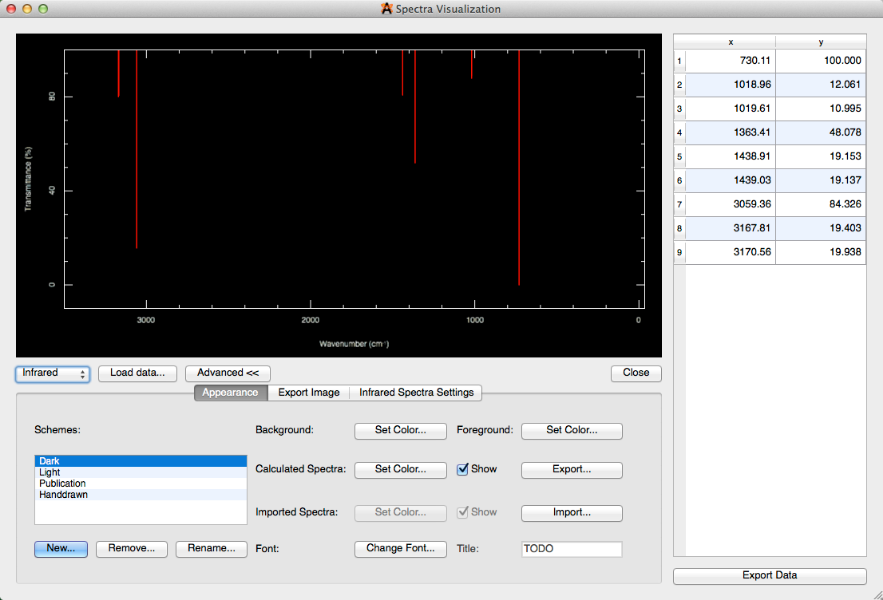
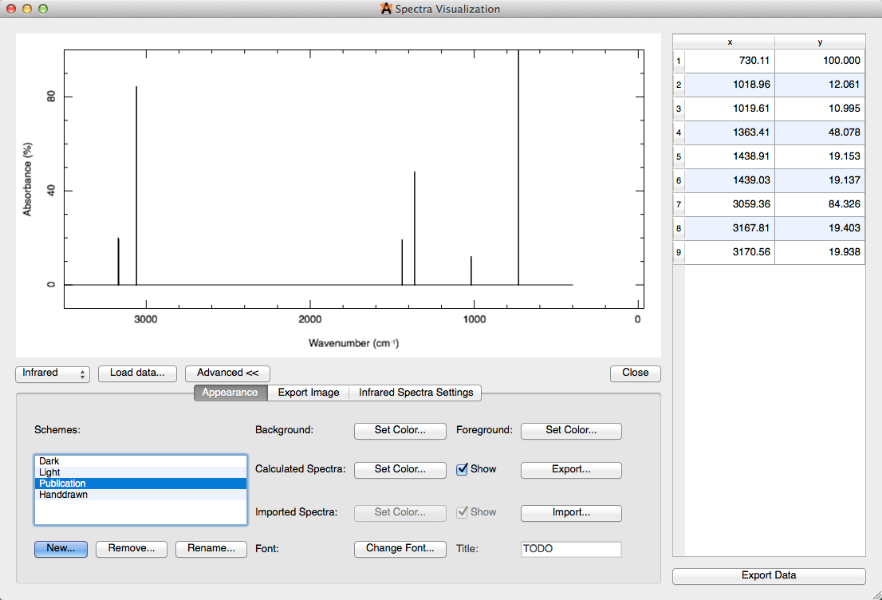
- Ejes
- Pelota y palo
- Dibujos animados
- Dipolo
- Fuerza
- Enlace de hidrógeno
- Etiqueta
- Polígono
- QTAIM
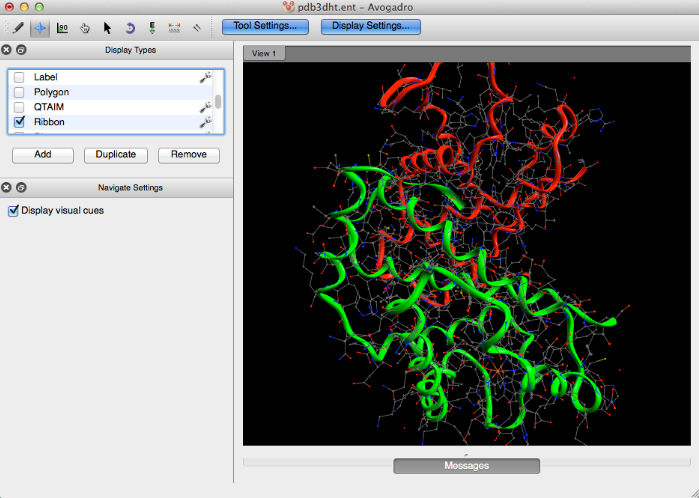
- Cinta
- anillo
- Estructura metálica simple
- Palo
- Superficie
- Van der Waals esferas
- Estructura de alambre
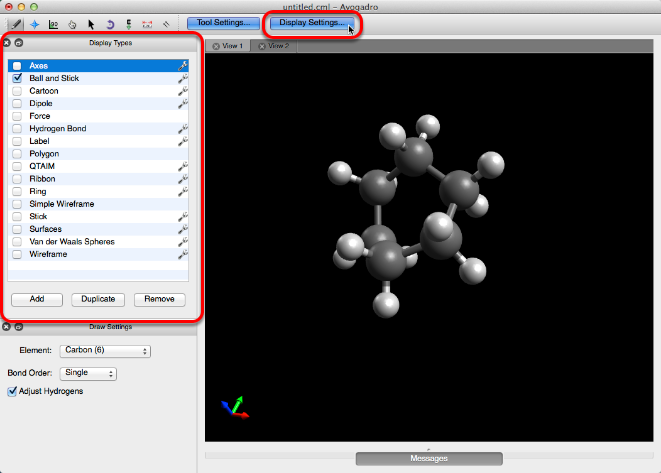
Localización de tipos de pantalla
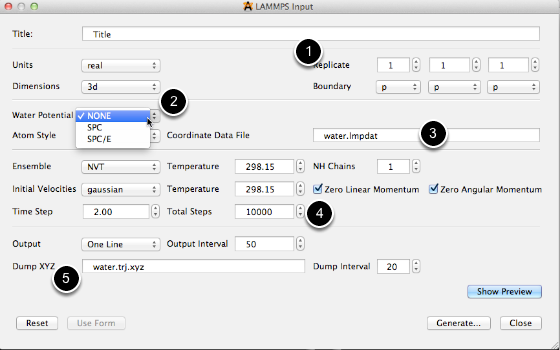
Se puede acceder a los diferentes tipos de pantalla haciendo clic en "Configuración de pantalla ..." en la parte superior central de la ventana abierta de Avogadro. La barra de herramientas de los tipos de visualización se agregará encima de la barra de herramientas "Configuración de herramientas ..." que está abierta actualmente. Todos los complementos que se muestran a continuación se pueden utilizar conjuntamente.
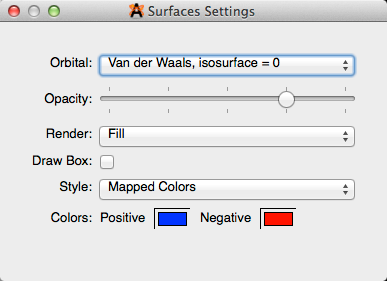
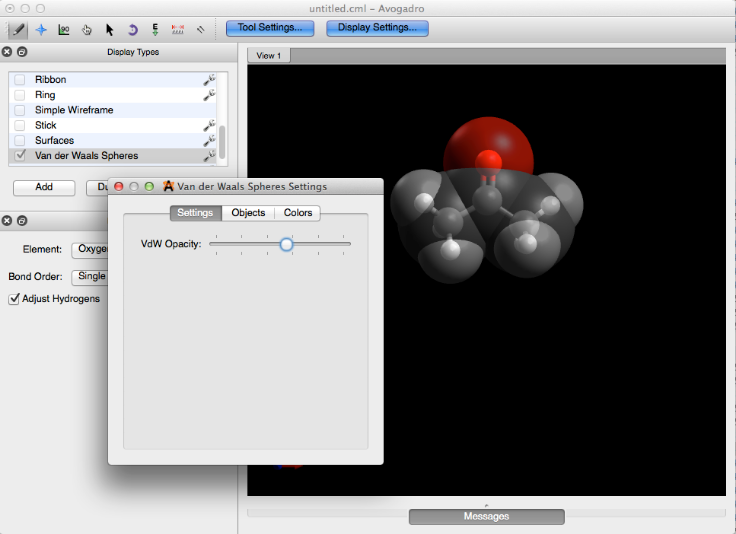
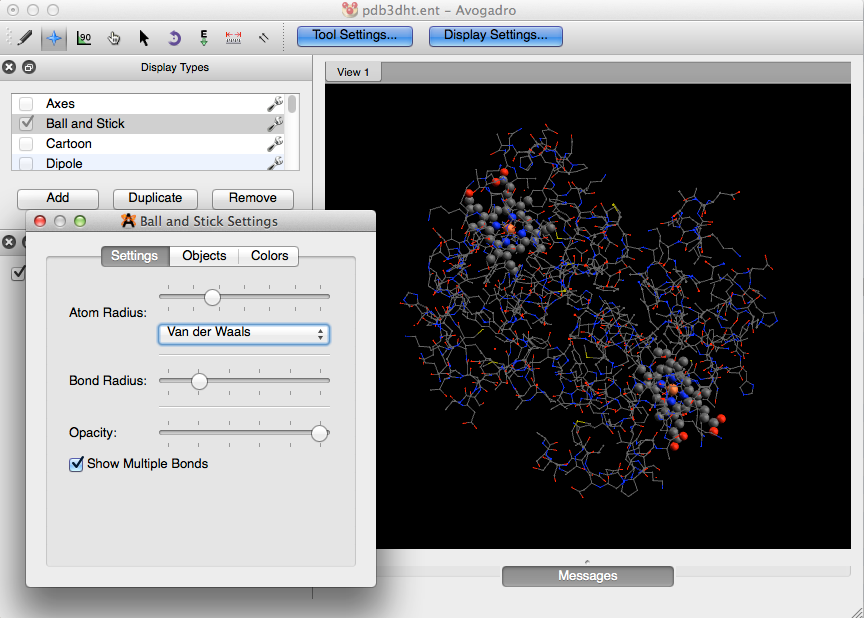
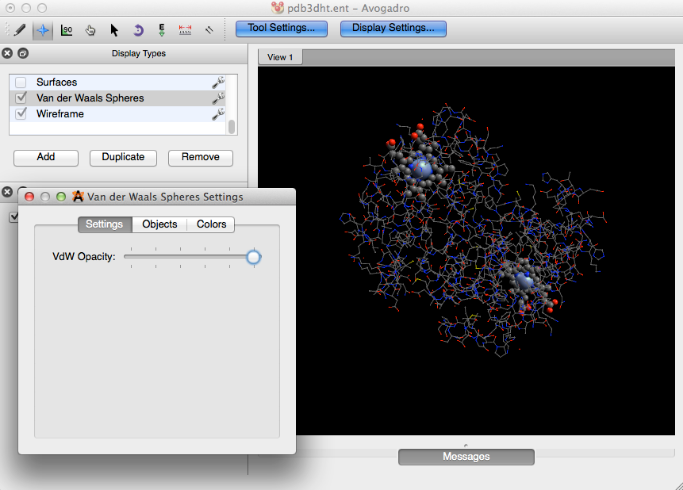
Al hacer clic en la llave ubicada a la derecha de algunos tipos de pantalla, se podrán realizar varios ajustes en la pantalla. Por ejemplo, puede optar por editar la opacidad de la función Esferas de Van der Waals para que aún pueda ver la bola y el modelo de palo debajo.
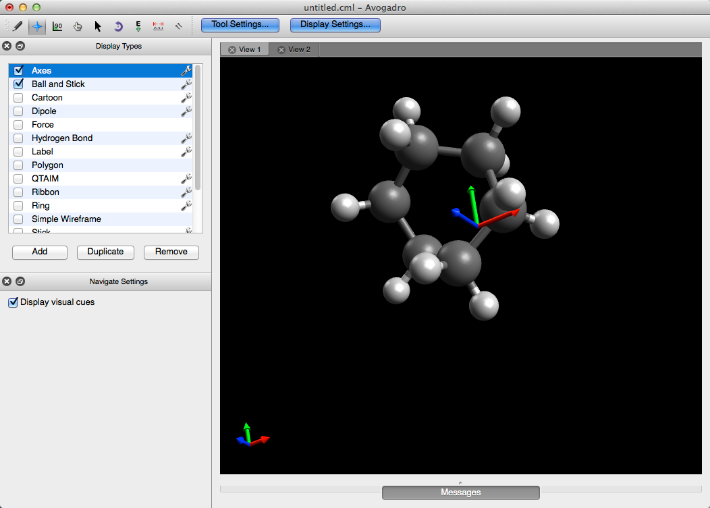
Ejes
Al hacer clic en el complemento Ejes proporcionará los ejes cartesianos de la molécula desde el origen. Tenga en cuenta que las flechas roja, verde y azul representan los ejes x, y y z, respectivamente.
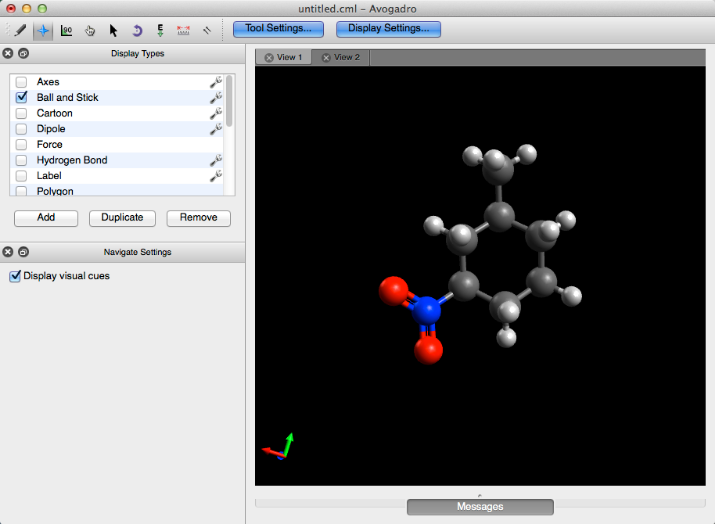
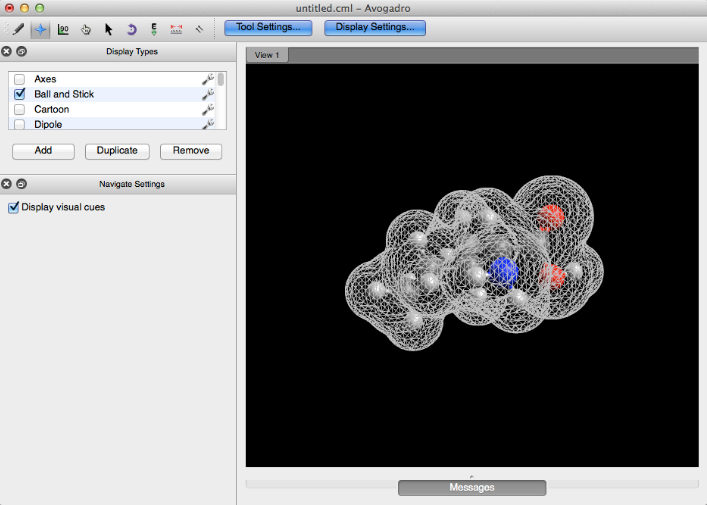
Pelota y palo
Ball and Stick es el complemento predeterminado cuando se abre Avogadro. Este complemento proporciona la representación estándar de bola y palo de una molécula.
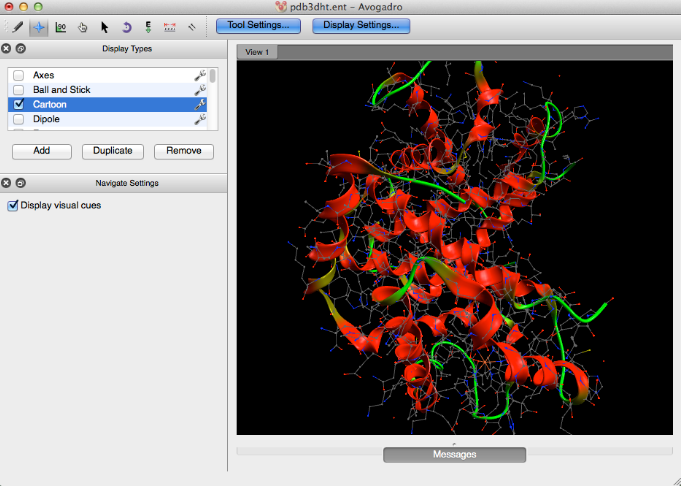
Dibujos animados
La función de dibujos animados solo se aplica a estructuras biológicas secundarias (hélice α y lámina β). A continuación se muestra la caricatura de hemoglobina.
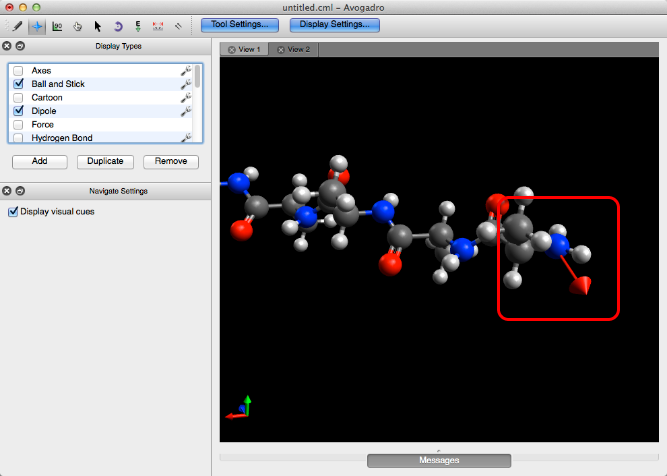
Dipolo
El complemento Dipolo mostrará un dipolo neto general si hay uno presente.
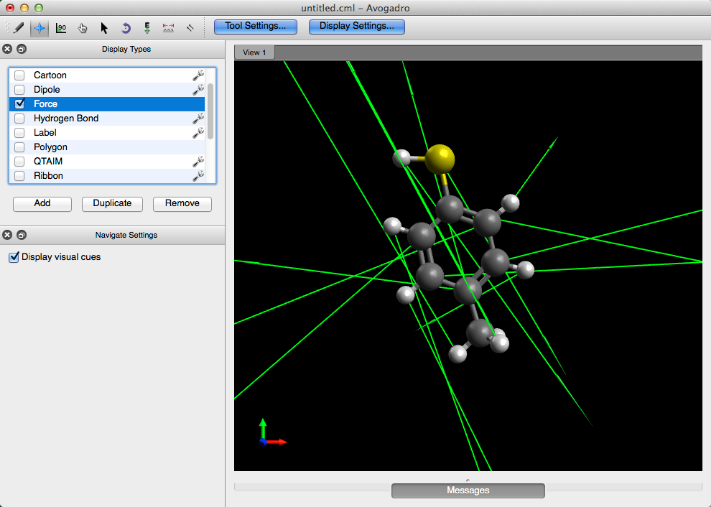
Fuerza
El plugin Force muestra flechas verdes en los átomos (como se muestra a continuación), para demostrar cualitativamente las fuerzas que se aplican a los átomos.
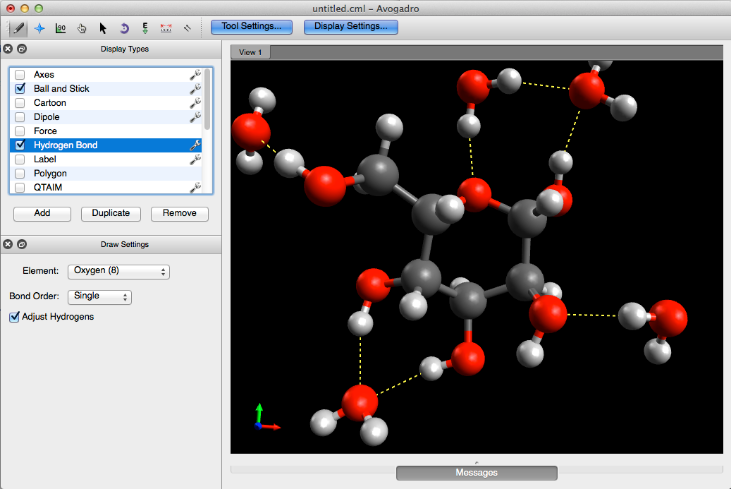
Enlace de hidrógeno
El plugin Hydrogen Bond demuestra los enlaces de hidrógeno implícitos que pueden ocurrir entre los átomos.
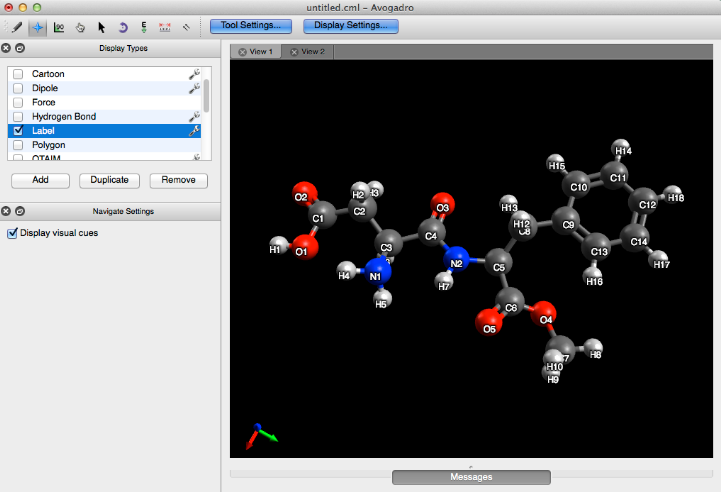
Etiqueta
La etiqueta numera el número y etiqueta todos los átomos presentes en una molécula.
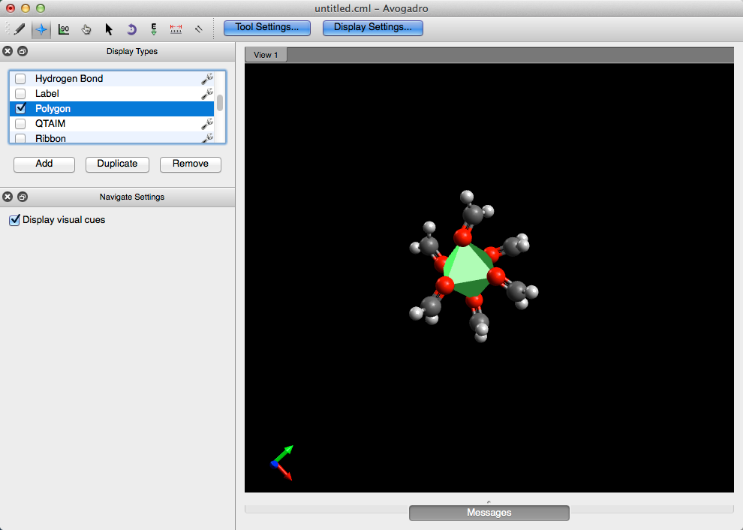
Polígono
La característica Polígono toma centros metálicos con tres o más átomos unidos a ellos, y dibuja un polígono alrededor de ellos.
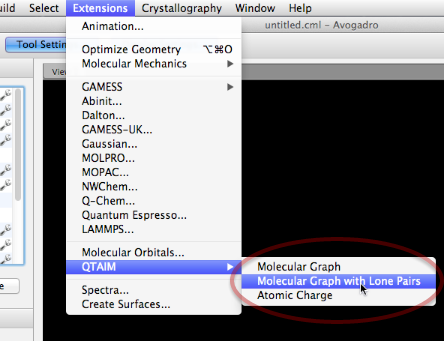
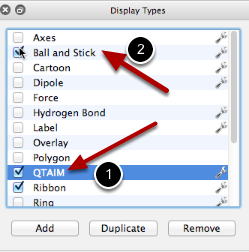
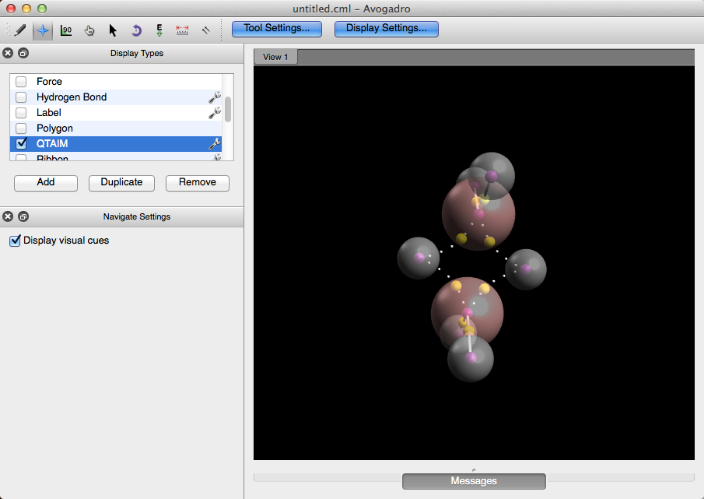
QTAIM (Teoría Cuántica de Átomos en Moléculas)
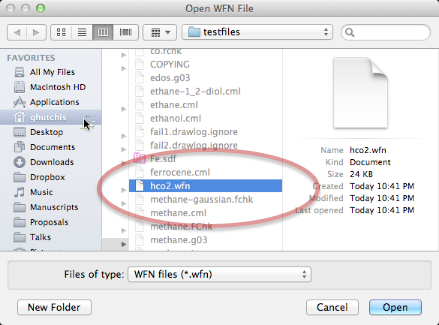
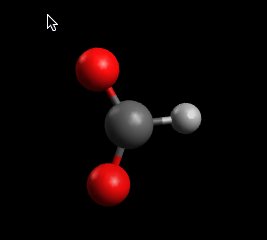
QTAIM muestra el enlace implícito que se teoriza para tener lugar entre los hidrógenos de los cristales orgánicos (el enlace implícito se transmite a través de puntos). Este tipo de pantalla se utiliza importando un archivo .wfn de la selección "QTAIM", "Molecular Graph" en el menú "Extensions". Puede encontrar más información sobre este proceso en la sección Tutorial de este manual.
Cinta
Al igual que el complemento Cartoon, el complemento Ribbon transmite estructuras biológicas secundarias como una simple representación de la cinta.
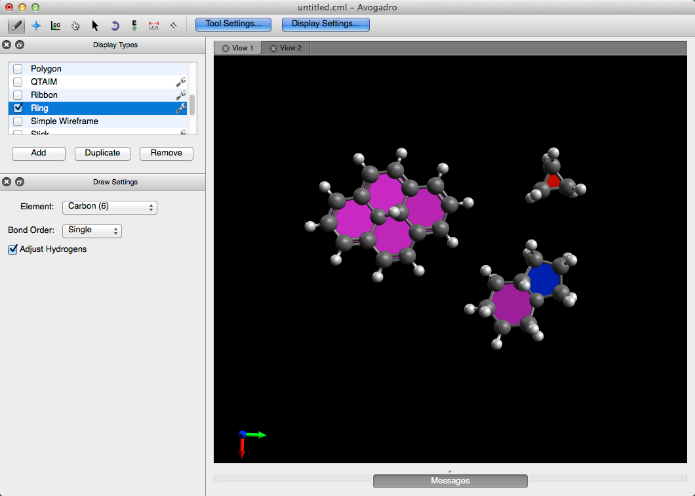
anillo
Esta característica distingue anillos con diferentes colores dependiendo de su tamaño. Como se muestra a continuación, un anillo de seis miembros es púrpura y el anillo de cinco miembros es azul, etc.
Estructura metálica simple
Esta característica proporciona una visualización básica de una molécula.
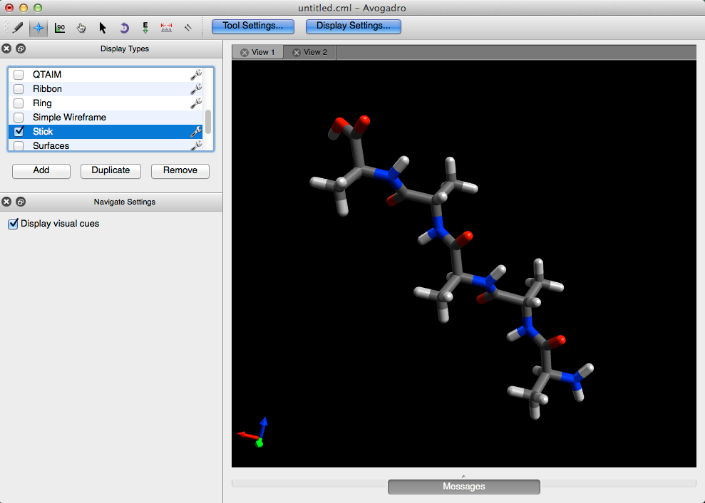
Palo
Stick es otro tipo de visualización molecular, que representa una representación en stick de una molécula.
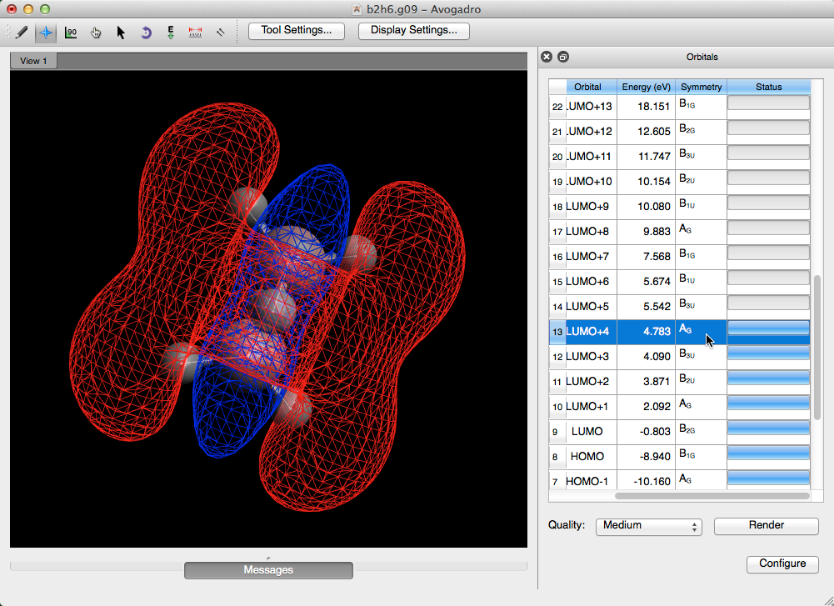
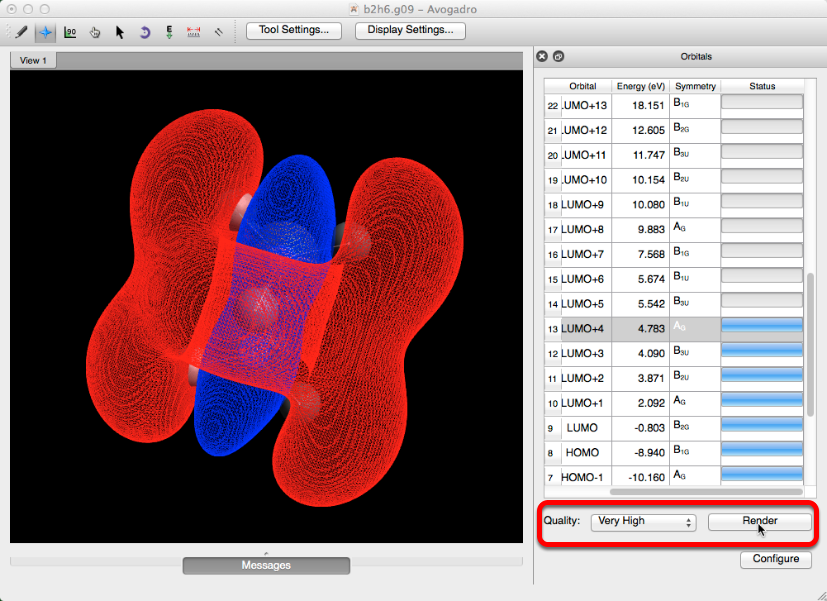
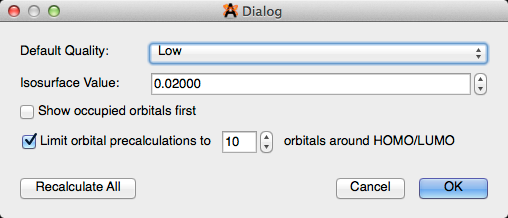
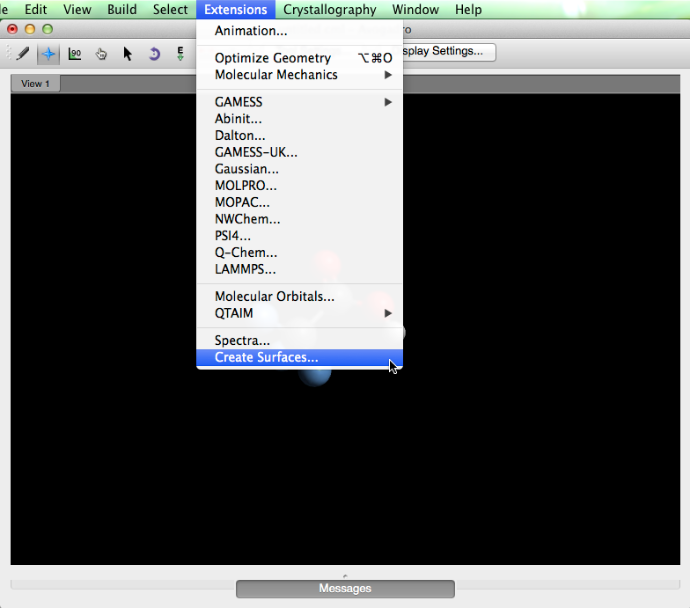
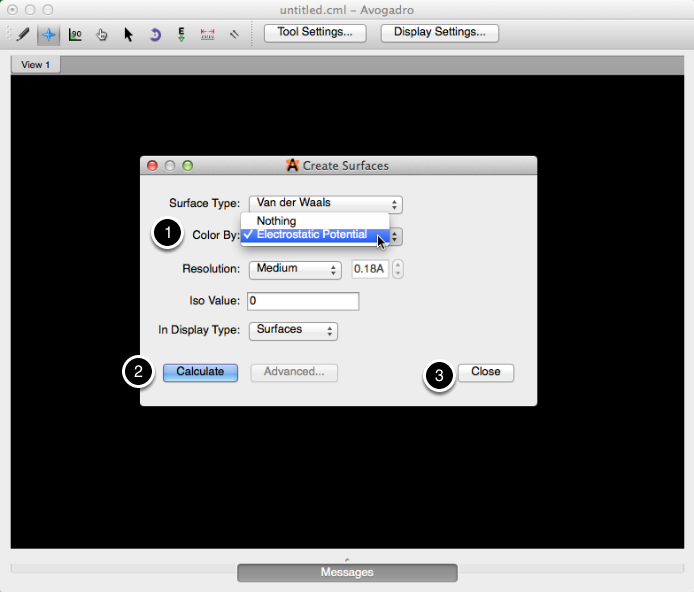
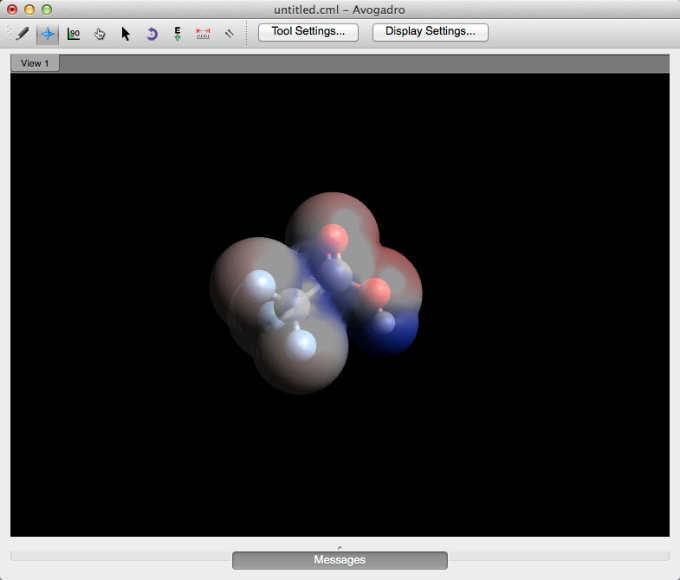
Superficies
Una vez que se ha creado una superficie (Menú de extensiones -> Crear superficies…), se puede utilizar el tipo de visualización de Superficie. Este tipo de pantalla permite ajustes en el orbital, opacidad, renderización, estilo y color.
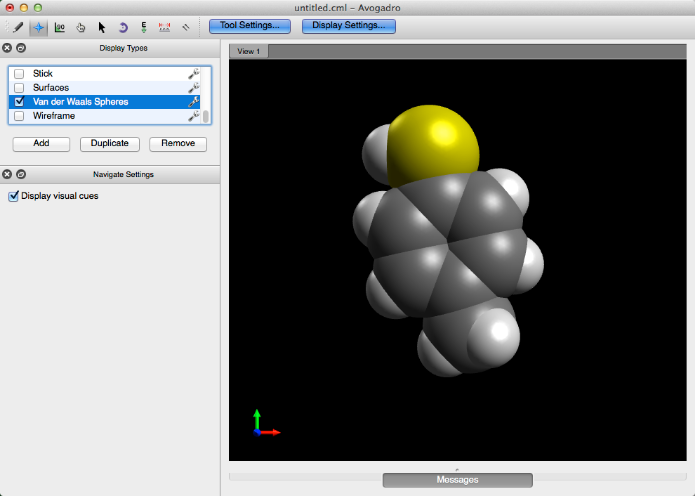
Van der Waals esferas
El complemento de Van der Waals proporciona la imagen de Van der Waals representada en la esfera clásica.
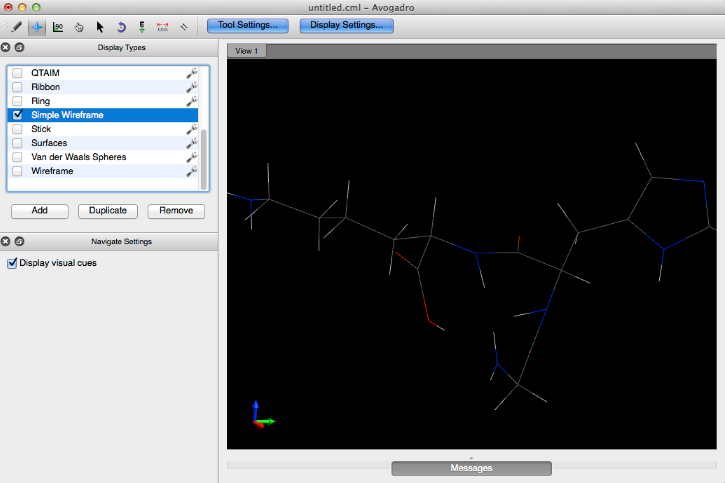
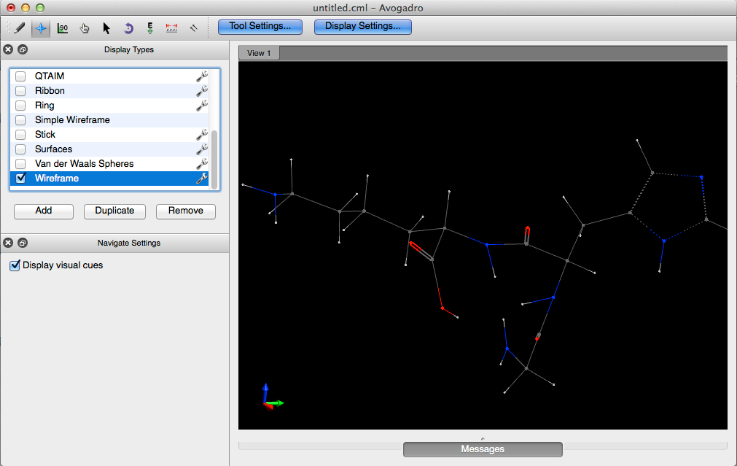
Estructura de alambre
A diferencia de Simple Wireframe, Wireframe dibuja átomos y un orden de enlace en la molécula.
Colorear parte de una molécula
Colorear varias partes de una molécula puede proporcionar una manera más visualmente estimulante de mostrar información cualitativa.
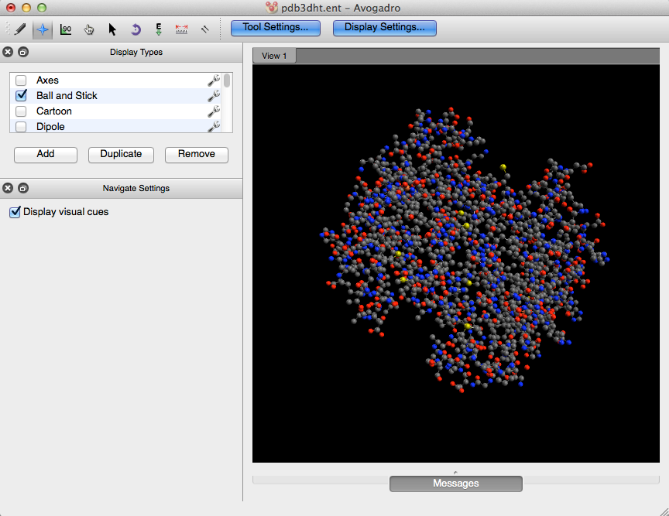
Hemoglobina
A continuación, se muestra la hemoglobina en su forma de bola y palo (archivo importado de PDB).
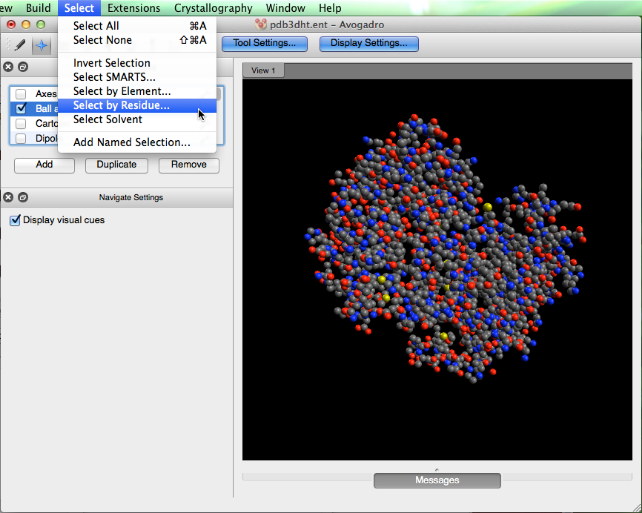
Seleccionar por Residuo
Podemos seleccionar residuos específicos en la molécula a través del menú "Seleccionar".
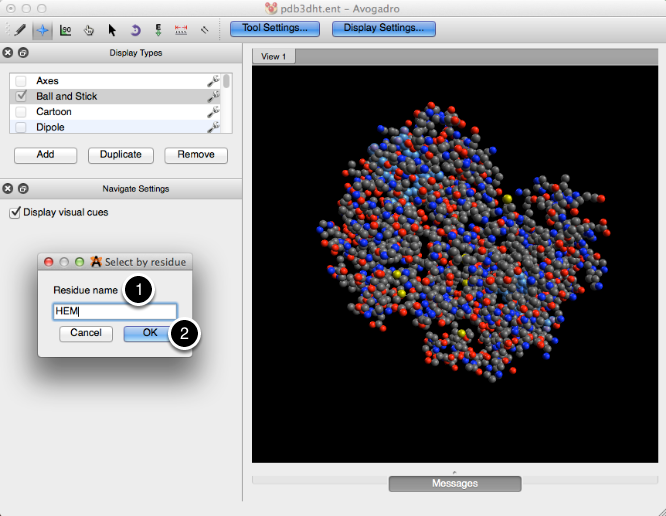
Después de escribir el nombre del residuo (esta función distingue entre mayúsculas y minúsculas) y hacer clic en "Aceptar", se pueden hacer ajustes para enfatizar la selección.
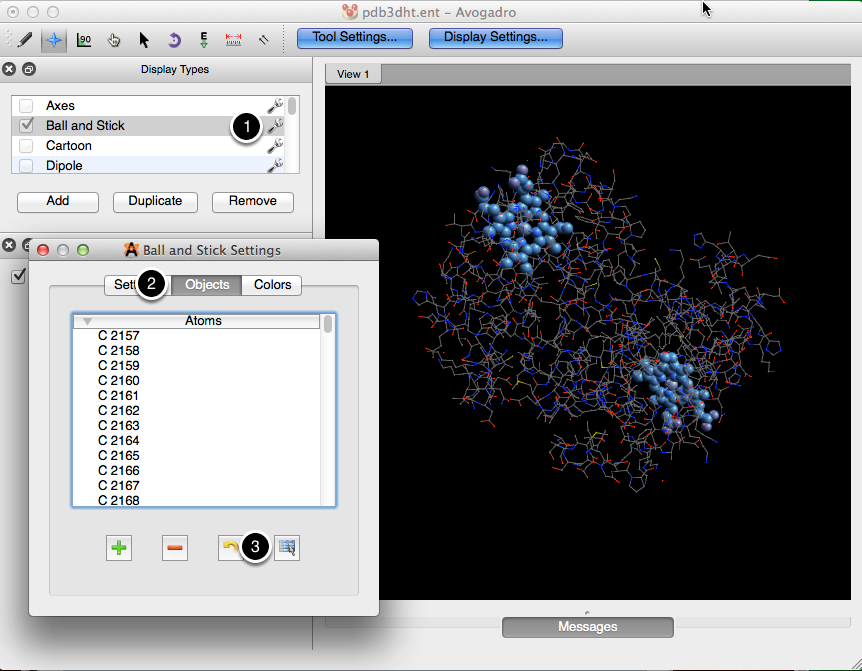
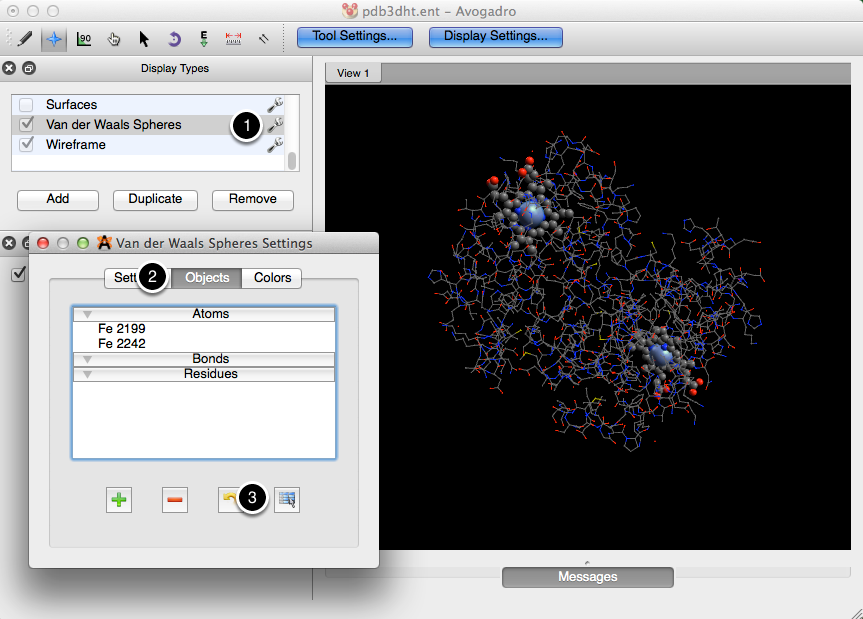
Después de hacer una selección, haga clic en la llave junto al tipo de pantalla que desea editar (en este caso, la pantalla de la bola y el palo). Cuando aparezca el cuadro de diálogo, seleccione "Objetos", y luego haga clic en el botón de la tabla azul en la esquina inferior derecha. Esta función ajusta lo que inicialmente se consideró un objeto (hemoglobina) y edita la selección para que el tipo de visualización solo abarque los residuos del hemo.
Desde allí puede editar otras configuraciones del tipo de pantalla haciendo clic en "Configuración" o "Colores".
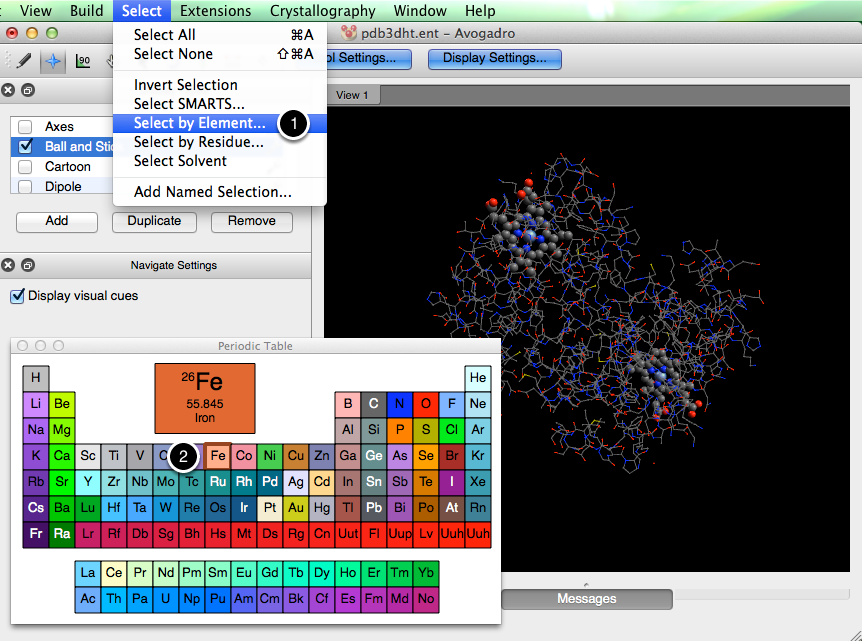
Seleccionar por elemento
El coloreado de las partes de una molécula no tiene que terminar después de un solo ajuste. Digamos que queremos enfatizar que el hierro está en el centro del residuo del dobladillo. Podemos ir al menú "Seleccionar", "Seleccionar por elemento ...", y elegir seleccionar todos los átomos de hierro en la molécula.
Una vez realizada la selección, con el mismo procedimiento que antes, podemos elegir un tipo de visualización y editar la selección para que solo abarque los átomos de hierro.
Luego se pueden hacer ajustes a la configuración.
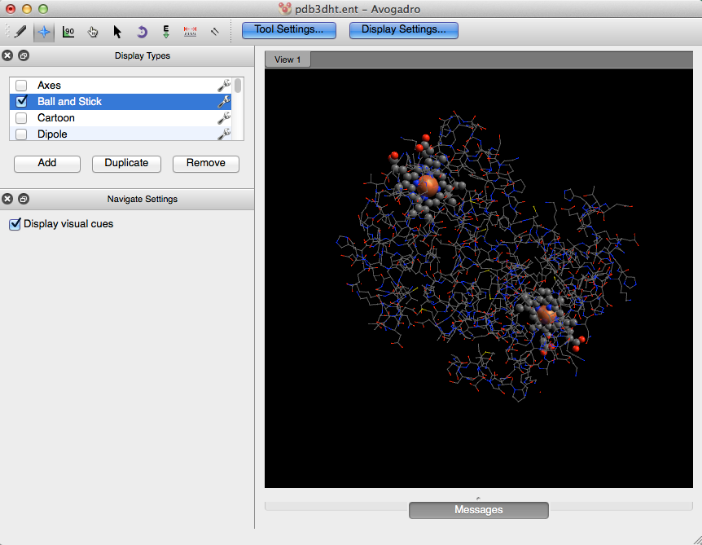
El producto final es una vista más intuitiva de la información cualitativa sobre la hemoglobina.