Super Cell Builder
Una vez que se ha construido una superficie de cristal, el Super Cell Builder puede expandir los átomos dentro de un grupo espacial, replicar la celda unitaria y realizar un enlace simple.
Cuando se selecciona "Super Cell Builder ..." en el menú "Crear", aparece el siguiente cuadro de diálogo. Este cuadro de diálogo le permitirá replicar una celda unitaria que ya se ha creado (si es necesario, se puede crear una celda unitaria seleccionando "Agregar celda unitaria" en el menú "Cristalografía").
Creando una superficie
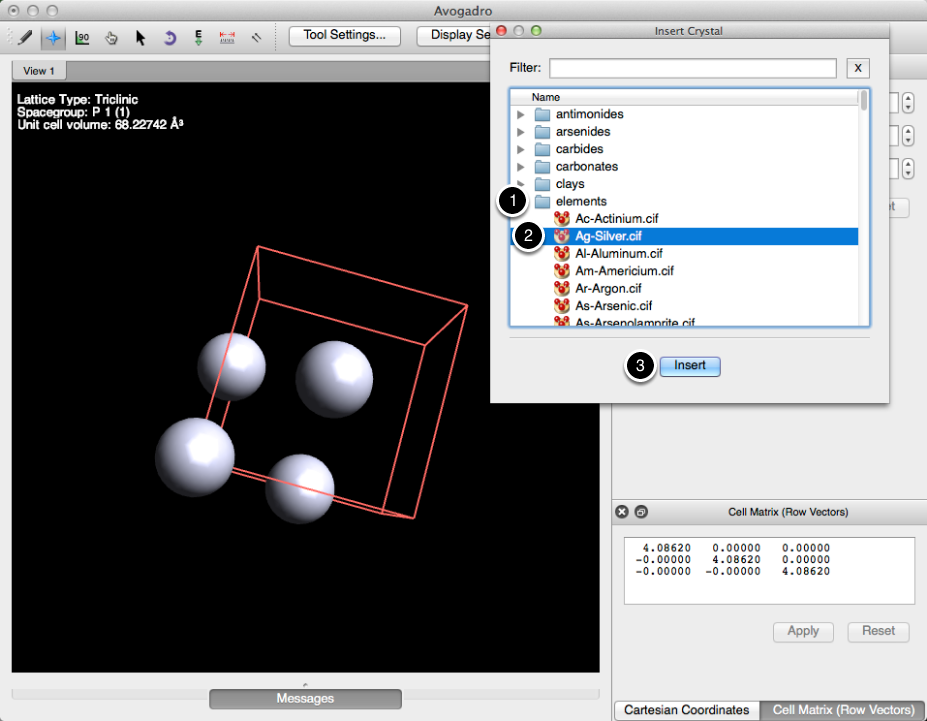
Una forma en que se puede utilizar una supercelda es creando una superficie. Abajo hay una celda elemental compuesta de plata. Esta celda se importó a través del menú "Archivo", en "Importar", "Cristal ...". Cuando aparezca el cuadro de diálogo, siga el procedimiento que se muestra a continuación.
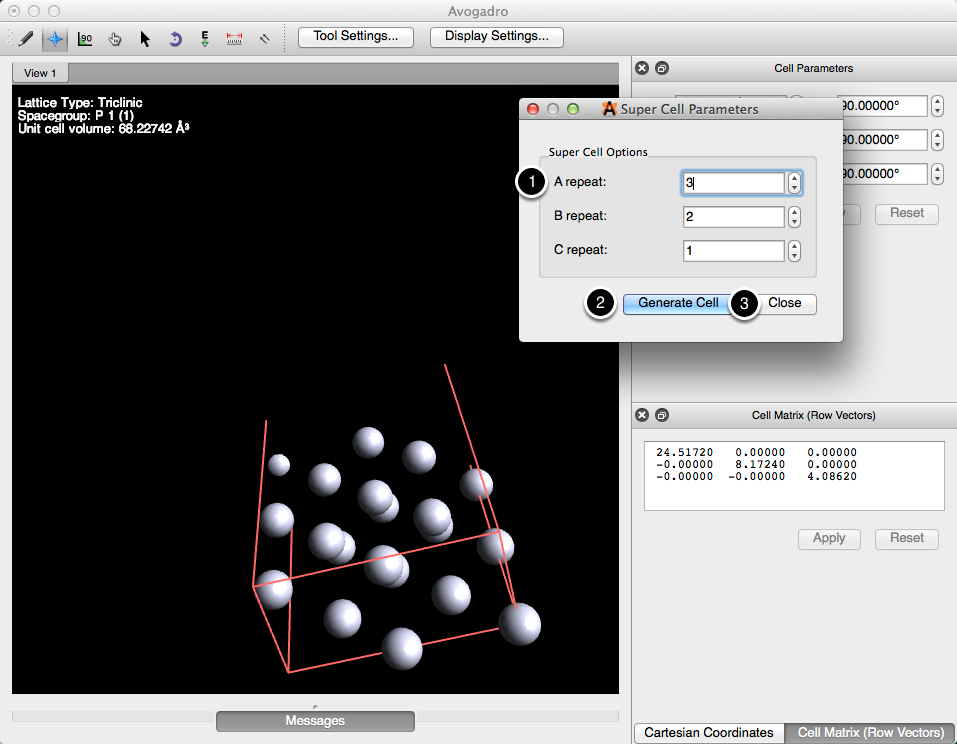
Una celda unitaria se puede replicar para hacer una * losa * o una superficie. Para este ejemplo, los parámetros se editaron como se muestra en la imagen a continuación. Después de editar los parámetros, haciendo clic en "Generar celda" expandirá su superficie.
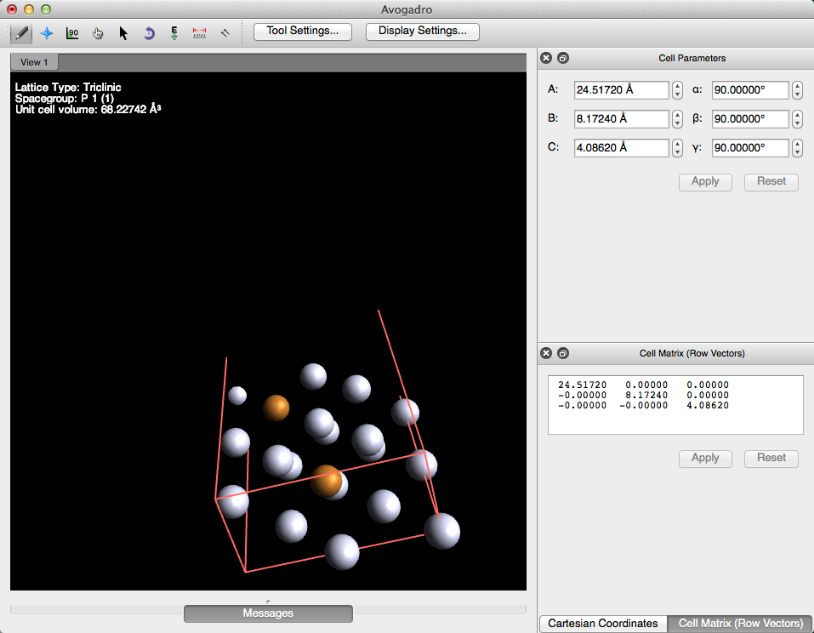
Una superficie puede ser modificada introduciendo impurezas. Aquí, las impurezas de cobre se añadieron a la superficie de plata. Este archivo ahora se puede exportar a otro programa para determinar, mediante cálculos, cómo las impurezas impactarán la superficie.
Construyendo una superficie de cristal (losa)
Construya una superficie de cristal, por ejemplo, Pt <111> para un plano de Miller definido.
Importar la estructura de cristal adecuada.
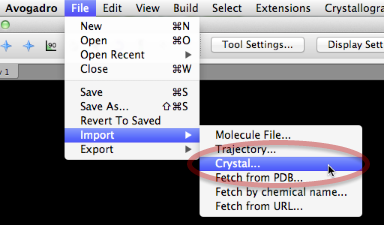
Abra un archivo CIF con la estructura de cristal necesaria o importe uno de la biblioteca de cristal Avogadro incorporada. El tutorial supondrá que importa una estructura desde la biblioteca de Avogadro. Elija Archivo> Importar> Crystal para abrir la biblioteca.
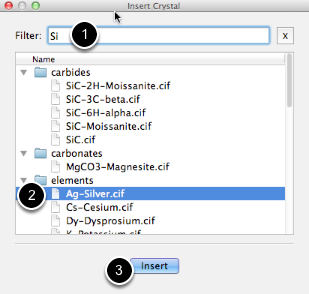
Navegue a través de los cristales o escriba un filtro, por elemento o nombre. Haga clic en "Insertar" para importar la estructura seleccionada.
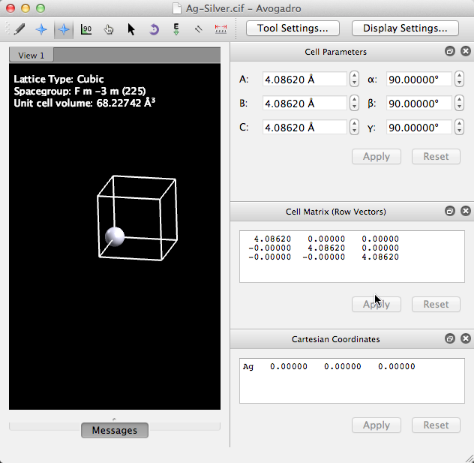
La importación de un cristal mostrará la celda unitaria asimétrica (por ejemplo, un átomo para Plata aquí).
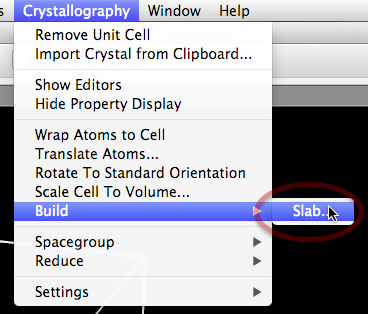
Para construir una superficie específica (por ejemplo, Ag <121>) elija Cristalografía> Construir> Losa ... para que aparezca la configuración del generador de losas. Los futuros constructores de cristales (p. Ej., Nanopartículas, supercélulas) también aparecerán en este menú.

Especifique los índices del plano de Miller deseado (para celdas de unidades hexagonales, aparecerán los 4 índices), y elija las dimensiones en distancias o celdas de repetición de la superficie resultante. La superficie generada se alinea en el plano XY, y un espesor específico se dividirá en el eje z debajo del plano XY. Esta característica permite una fácil alineación entre una nueva superficie y una molécula para los cálculos de interacción de superficie. Haga clic en "Crear" para iniciar la generación de superficie.
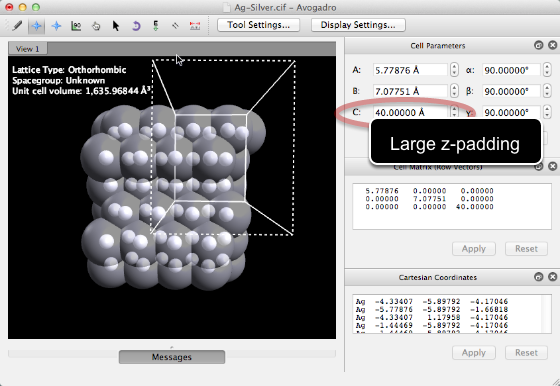
Después de hacer clic en "Crear", Avogadro generará una gran supercélula, alineará, rotará y dividirá la superficie designada. Esto puede llevar algún tiempo, dependiendo del tamaño de la célula de cristal. Aquí se utilizan esferas translúcidas de van der Waals para ilustrar la ondulación de la superficie de Ag <121>. La superficie resultante es una supercélula de 2x2, con un espaciado grande (40 Å) en el eje z.
Construyendo una célula de unidad de polímero
Un recorrido por la creación de una celda unitaria (de un polímero) utilizando Avogadro y la herramienta Alinear. Este ejemplo específico usa gaussiano, pero los vectores de traducción para otros programas se pueden realizar de manera similar.
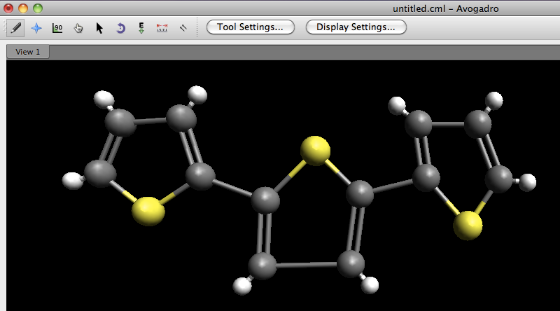
Construye la molécula para la célula unitaria. Tenga en cuenta que aunque la unidad de repetición aquí es de 2 timbres, hemos construido 3 timbres. De esta manera, modelaremos correctamente el enlace que abarca dos celdas unitarias.
Optimizar la geometría de la molécula.
Cambie a la herramienta Alinear para traducir y orientar las coordenadas de la celda unitaria.
Asegúrese de abrir la ventana Configuración de herramientas, que le permitirá trabajar con la herramienta Alinear.
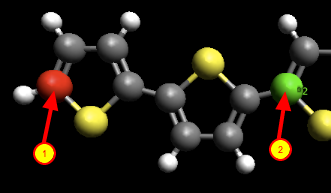
Primero haga clic en el átomo de "inicio" del politiofeno. Este átomo será traducido al origen (0, 0, 0). Luego haga clic en el átomo correspondiente en la celda de la unidad "siguiente". La distancia entre estos dos átomos definirá un eje en la celda unitaria.
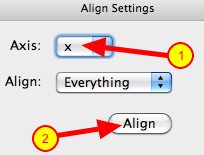
En la ventana "Alinear configuración", defina un eje para la celda unitaria. Luego haga clic en el botón Alinear. Esto cambiará el conjunto de coordenadas para tener el átomo # 1 en el origen, y el átomo # 2 (desde el paso anterior) proyectado en el eje x.
Abra la ventana del editor cartesiano para verificar los resultados de la operación de alineación.
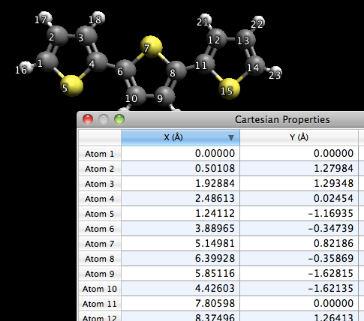
Observe que el átomo # 1 está en el origen y el átomo # 11 se proyecta en el eje X. El tamaño de la celda unitaria es 7.806Å, la distancia entre el átomo # 1 y el átomo # 11.
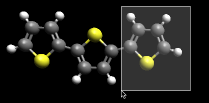
Ahora elimine los átomos "adicionales" que no deben incluirse en los cálculos de la unidad de la célula. Esto incluye el tercer anillo (incluido el átomo 11) y los átomos de hidrógeno "finales". Por ejemplo, puede usar la herramienta de selección y arrastrar sobre los átomos que se eliminarán para seleccionarlos.
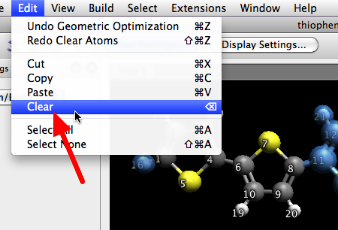
Una vez seleccionado, puede utilizar el comando de menú "Borrar" para eliminar los átomos.
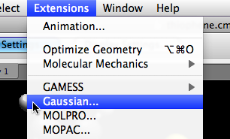
Si desea enviar la celda unitaria a gaussiano, seleccione la extensión de entrada gaussiana.
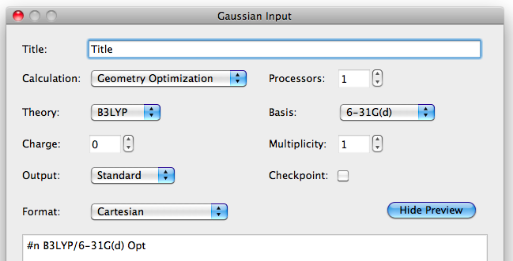
Establece las opciones que desees. Asegúrese de agregar una línea de “TV 7.806 0.0 0.0” en la parte inferior del texto de vista previa. Esto permitirá el cálculo de la celda unitaria configurando el vector de traslación para la celda unitaria.
Percepción de simetría de cristal
Los resultados del cálculo a menudo especifican todos los átomos y vectores de traducción, pero no el grupo espacial. Aquí vemos cómo percibir el grupo espacial desde un conjunto de coordenadas cristalográficas.
Abrir un archivo de cristal
Aquí abrimos un ejemplo de cálculo VASP abriendo el archivo POSCAR.
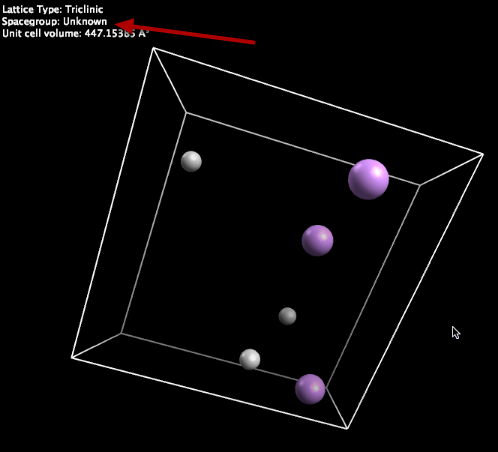
Este ejemplo es triclínico, buscando estructuras de L .. Tenga en cuenta que los archivos VASP no especifican un grupo de espacio, por lo que se informa como "Desconocido".
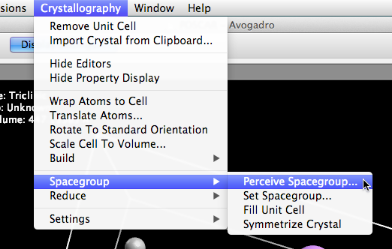
Podemos establecer el grupo espacial en forma manual, o aquí, percibir el grupo espacial, usando el código de código abierto de spglib.
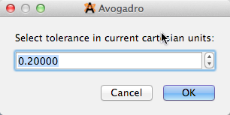
Necesitamos establecer la tolerancia, ya que algunos átomos pueden estar ligeramente fuera de lugar en las coordenadas cartesianas.
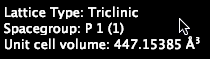
Nuestro ejemplo de archivo VASP no es muy interesante, el grupo espacial es P1.
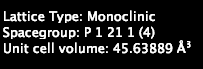
Aquí hay otro ejemplo, donde el grupo espacial es P 1 21 1.
Reducir los cristales a células unitarias primitivas
Algunas simulaciones utilizan “supercélulas”: sistemas de límites periódicos más grandes que la celda unitaria primitiva. Aquí hay un tutorial sobre cómo reducir una supercélula grande a la celda unitaria primitiva.
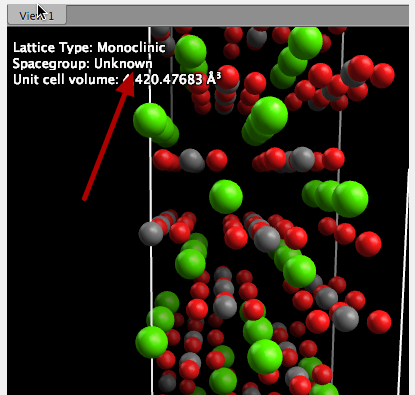
Abra o importe el archivo con la supercélula, aquí, CaCO3. Tenga en cuenta que el grupo de espacio es desconocido, ya que el archivo proviene de VASP, que no especifica un grupo de espacio con las coordenadas.
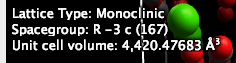
Después de percibir el grupo espacial, vemos correctamente que el sistema es R -3 c. Ahora podemos reducir la supercelda a una célula primitiva de CaCO3.
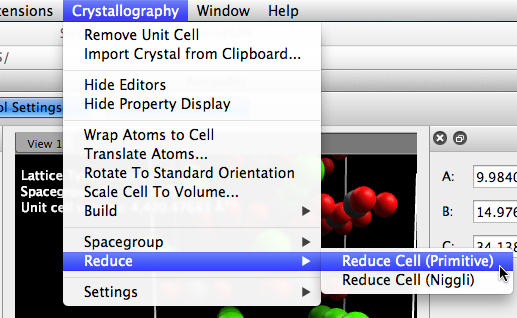
Avogadro proporciona dos algoritmos para reducir la celda unitaria a una celda primitiva o Niggli. Aquí, seleccione "Primitivo". Tenga en cuenta que el volumen de esta supercélula era superior a 4,000 Å3.
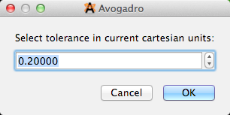
Deberá establecer una tolerancia para las coordenadas cartesianas (aquí, en Å).

Después de la reducción, tenga en cuenta que el grupo de espacio se conserva, la red es correctamente romboédrica y el volumen de la celda unitaria es 36 veces menor.
Volúmenes de cristal de escala
Avogadro 1.1 le permite ajustar el volumen o el espaciado de una celda unitaria.
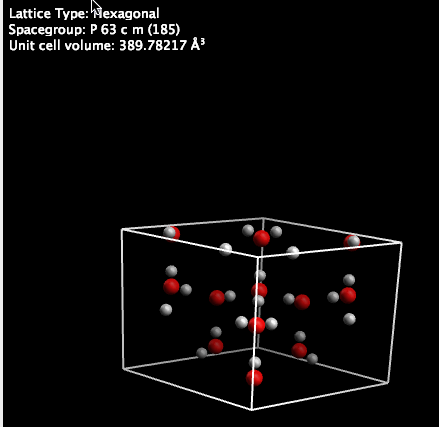
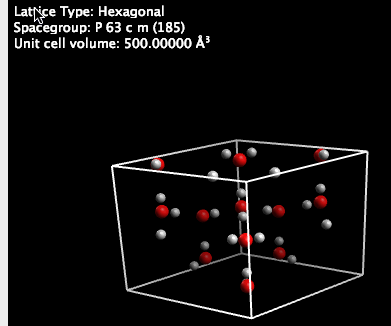
Después de crear o abrir el cristal (aquí el hielo), vemos la celda normal y la información de la red. Ahora ajustaremos el volumen de la celda.
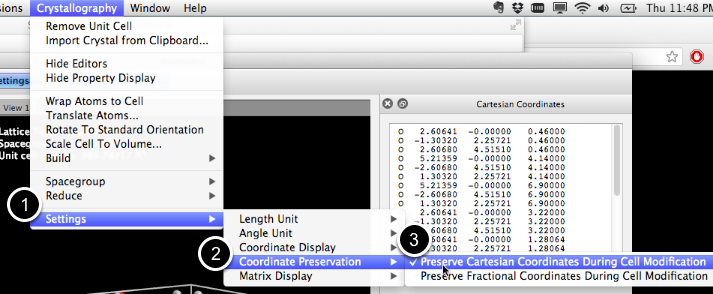
Antes de escalar el volumen, podemos optar por modificar las coordenadas cartesianas (que agregarán espacio vacío a los bordes de la celda unitaria) o conservar las coordenadas fraccionarias (que escalarán simétricamente la celda unitaria completa). Este recorrido mostrará ambos.
Primero escalaremos la celda conservando las coordenadas cartesianas.
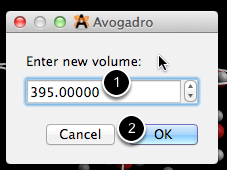
Las unidades del volumen están determinadas por su configuración (aquí Å). Ajustamos el volumen del original 389.78Å3 y hacemos clic en "Aceptar".
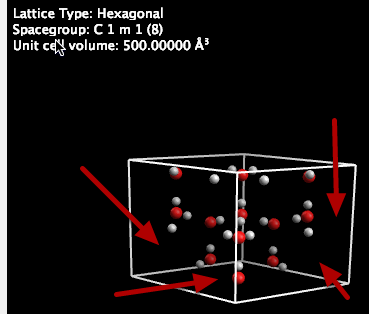
Aquí, hemos exagerado enormemente el volumen para mostrar el espacio vacío (flechas) alrededor del exterior de los límites de la celda unitaria, al preservar las coordenadas cartesianas. El grupo espacial también ha cambiado (a C 1 m 1).
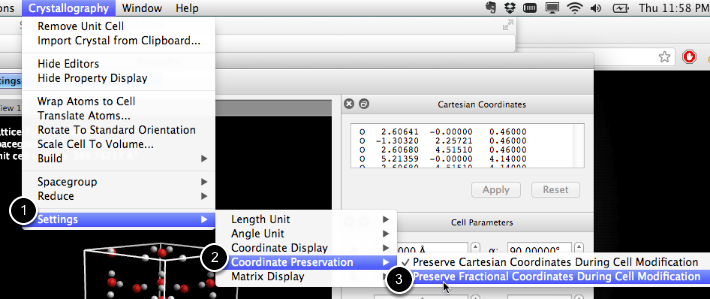
Si conserva las coordenadas fraccionarias, puede escalar la celda unitaria de forma simétrica.
Tenga en cuenta que mientras el volumen se expande significativamente, el grupo de espacio (y las coordenadas fraccionarias) se mantienen.
Interacciones Molécula-Superficie
Más allá de la construcción de una superficie de cristal, las nuevas características de Avogadro facilitan la consideración de las interacciones entre la molécula y la superficie. La lección comienza al final de la lección "Construyendo una superficie de cristal".
Comience con una superficie de cristal generada
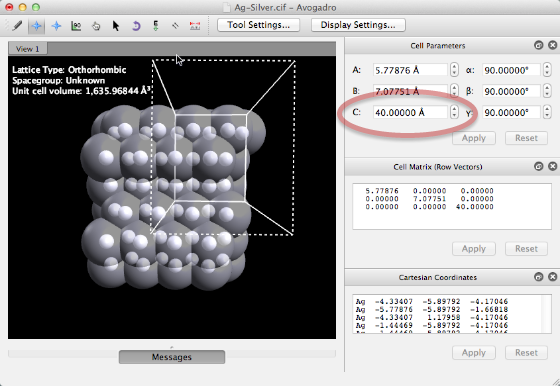
Generar la superficie de cristal deseada. Avogadro centrará la celda de la superficie, alineada en el plano XY, con los átomos de losa definidos por debajo de Z = 0. El Generador de losas también deja un gran espacio a lo largo del eje z para permitir la inserción de moléculas en los cálculos de interacción de la superficie. Puede controlar este relleno como se indica arriba.
Nueva ventana: crea nuestra molécula
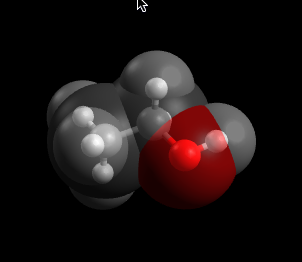
En una nueva ventana, dibuje la molécula deseada o abra un archivo. Aquí consideramos el etanol.
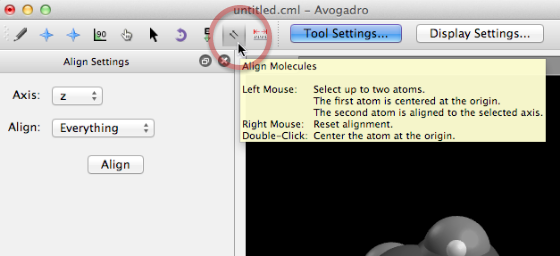
Usaremos la "Herramienta de alineación" para permitirnos rotar y alinear la molécula con el grupo OH en el origen y la molécula alineada a lo largo del eje z.
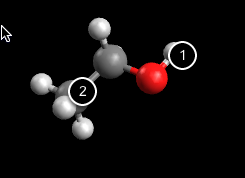
Haremos clic en el átomo H terminal (que se traducirá al origen) seguido del átomo de carbono (que definirá el eje z de la molécula).
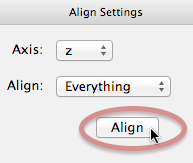
Después de definir los átomos (mostrarán esferas de colores y números una vez seleccionados), haga clic en el botón "Alinear" para traducir y rotar la molécula.
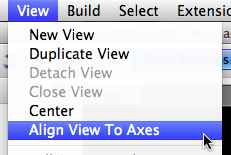
Es posible que desee modificar la vista actual de la cámara. Si selecciona Ver> Alinear vista con ejes, la vista se reiniciará para proyectar el eje z de la molécula para que apunte hacia usted.
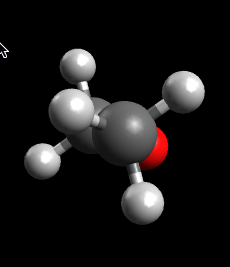
¡Perfecto! Ahora podemos copiar nuestro etanol al documento de superficie.
Después de copiar, podemos cambiar a nuestra superficie.
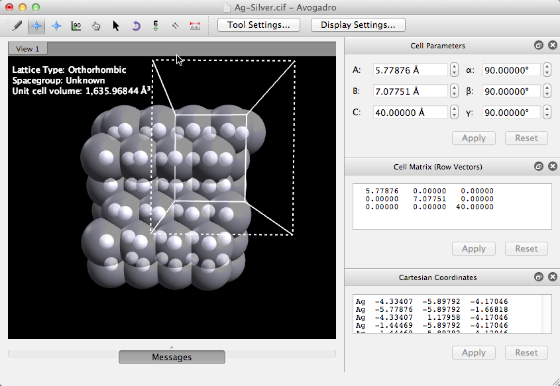
Ahora vamos a pegar en la molécula de etanol.
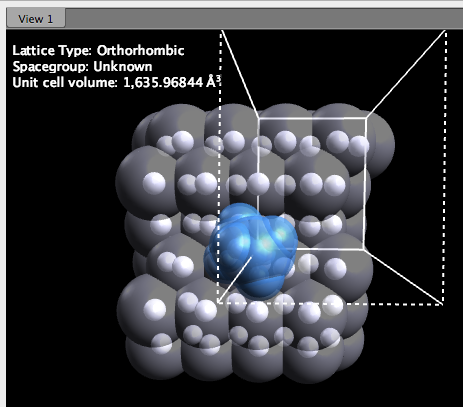
Tenga en cuenta que el etanol ahora está incrustado en la superficie, centrado como se desee. La herramienta Manipular ha sido seleccionada, nos permite traducir la molécula según sea necesario.
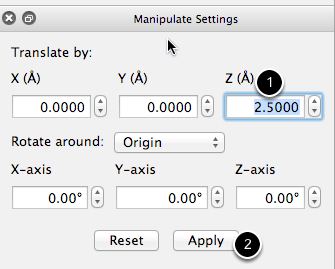
Nueva en la versión 1.1 es una opción para especificar la cantidad exacta para traducir o rotar la selección (es decir, la molécula que acabamos de pegar). Aquí, hemos especificado que queremos mover la molécula + 2.5Å a lo largo del eje z, sobre la superficie, y luego hacemos clic en "Aplicar" para completar. También podríamos girar alrededor del eje z si la posición no es la deseada.
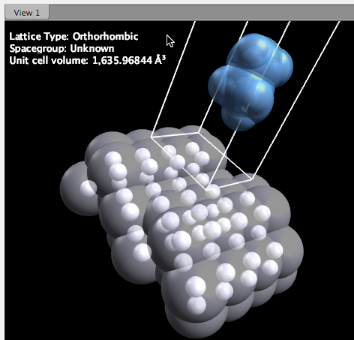
Aquí hemos traducido el etanol 2.5 Å por encima de la superficie Ag <121> y estamos listos para enviarlo para un cálculo.
No hay comentarios:
Publicar un comentario