Nombrando una molécula con PubChem
Avogadro 1.1 y versiones posteriores incluyen soporte para nombrar compuestos utilizando el sistema NIH Chemical Resolver y la base de datos PubChem.
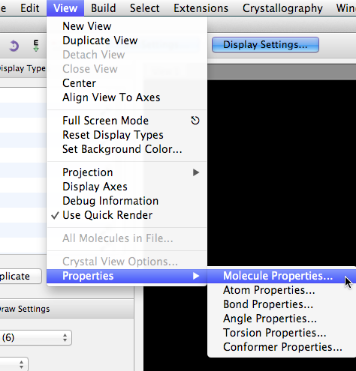
Abra la ventana Propiedades de la molécula, en el menú Ver.
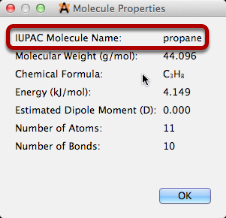
El nombre de IUPAC se mostrará inicialmente como (pendiente) mientras el servidor devuelve el nombre, (desconocido) si la molécula no se encuentra en el resolvedor, o (no disponible) si su conexión de red no funciona o el servicio de resolución no está disponible.
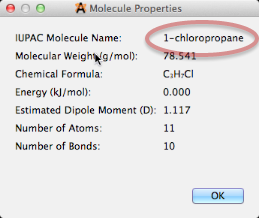
A medida que modifique la molécula (por ejemplo, agregando un átomo de Cl), el nombre se actualizará automáticamente con las otras propiedades.
Viendo los orbitales moleculares
Esta función requiere un "punto de control" o "punto de control con formato" de los códigos de química cuántica
Cuando se abre el archivo de salida, si se encuentra un archivo de punto de control coincidente, se abre automáticamente la barra de herramientas Orbital.
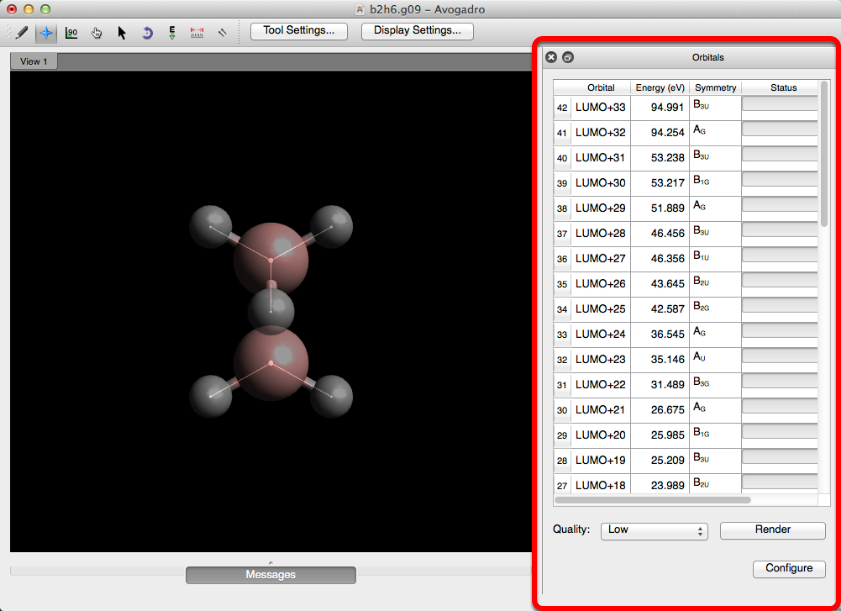
Todos los orbitales moleculares potenciales tendrán barras de estado completas (es posible que deba desplazarse hacia abajo considerablemente para encontrar los orbitales potenciales). Al hacer clic en la fila de un orbital, con una barra de estado completa, se creará una representación rápida y de baja calidad del orbital.
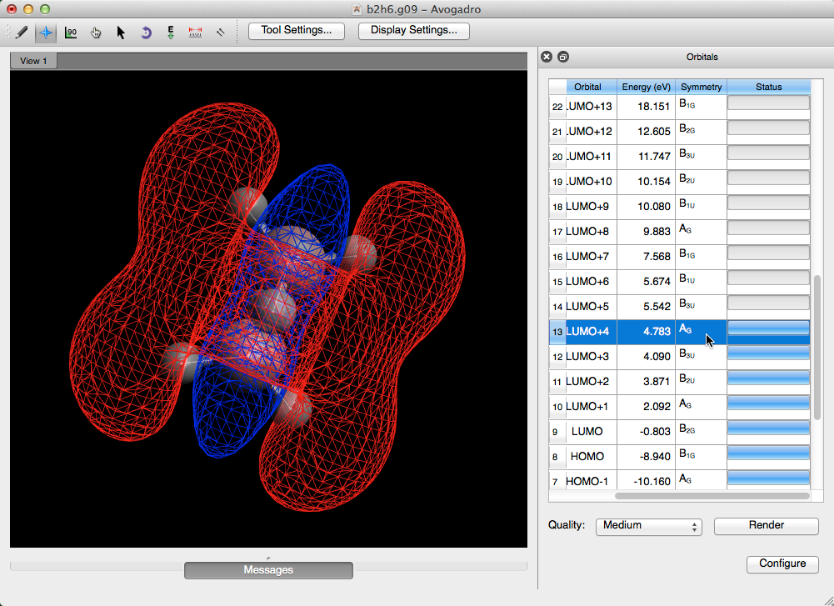
Si se desea, se puede seleccionar y aplicar una calidad orbital más alta. Esto se hace seleccionando una nueva calidad de imagen en el menú desplegable y haciendo clic en "render". Estas versiones pueden tardar un momento en cargarse.
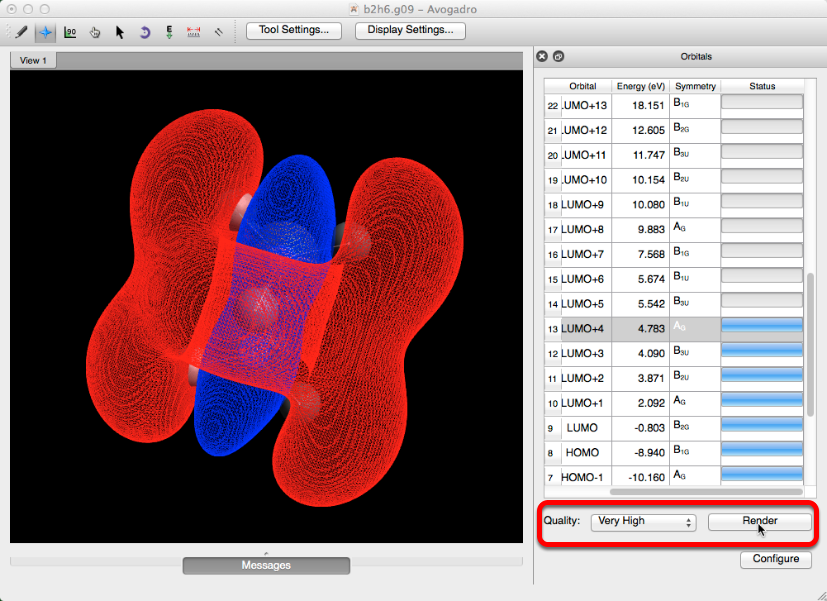
Configurar
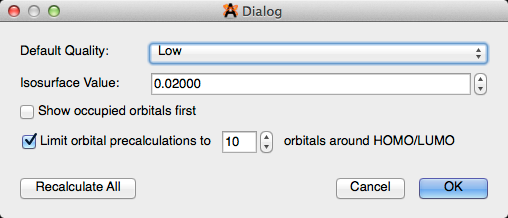
Seleccionar "Configurar" le permite ajustar los parámetros predeterminados para la barra de herramientas orbital. Una vez que los parámetros se hayan ajustado, haga clic en "Recalcular todo" antes de cerrar el cuadro de diálogo. "Recalcular todo" reevalúa y actualiza todos los parámetros.
Visualización de mapas de potencial electrostático
Los mapas de potencial electrostático ayudan a visualizar la distribución de carga y otras propiedades de las moléculas relacionadas con la carga.
En general, digamos que desea determinar visualmente si un protón específico tiene más o menos densidad de electrones. Primero, querrá comenzar con la molécula de su elección (a continuación se muestra el ácido trifluoracético).
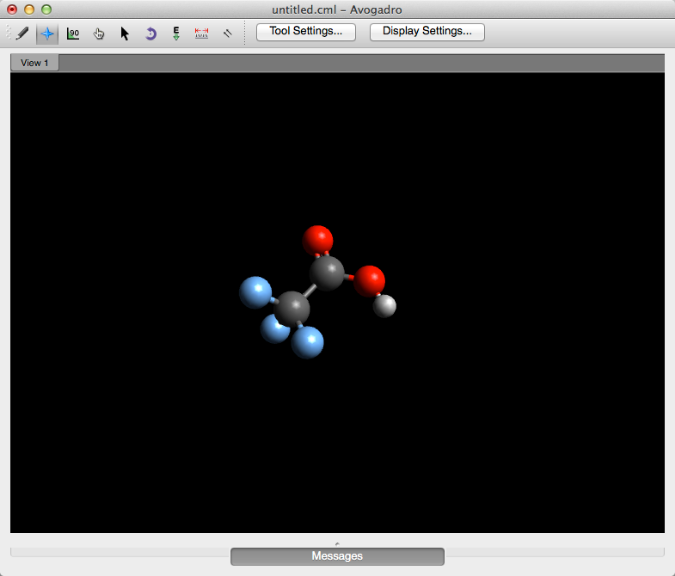
Luego, en el menú "Extensiones", seleccione "Crear superficies ...".
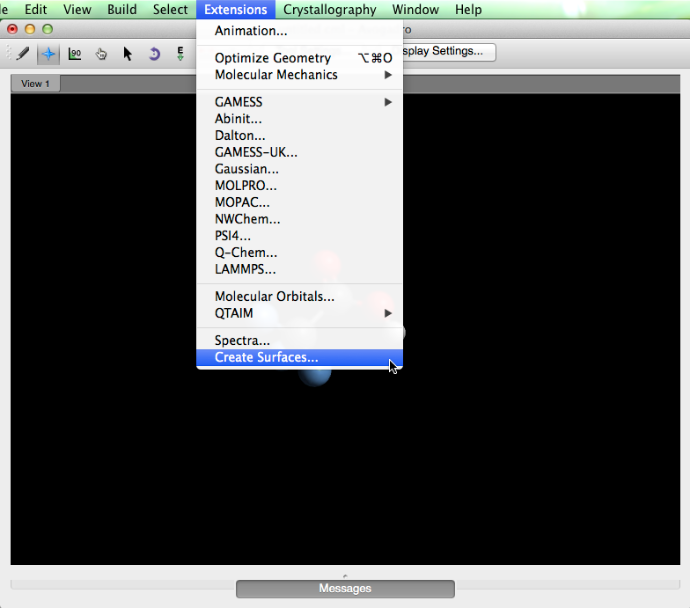
Aparecerá un cuadro de diálogo que le proporcionará varias opciones de superficie. En "Color por:", seleccione "Potencial electrostático", y luego haga clic en "Calcular". Después de que Avogadro calcula la superficie, seleccione "Cerrar".
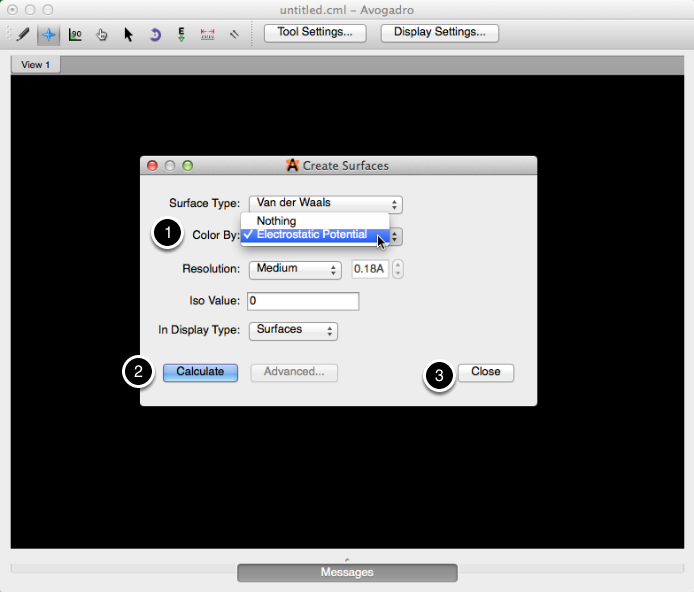
Se ha creado una superficie electrostática. Desde esta superficie, puede interpretar dónde reside la mayor densidad de electrones (en las áreas más rojas) y donde reside la menor densidad de electrones (áreas azules profundas). Puede determinar y comparar la acidez de varios protones, y cómo los átomos circundantes impactan la densidad electrónica global.
Este ejemplo fue tomado de "Explorando la acidez de las moléculas orgánicas con Avogadro" escrito por Tamika Madison.
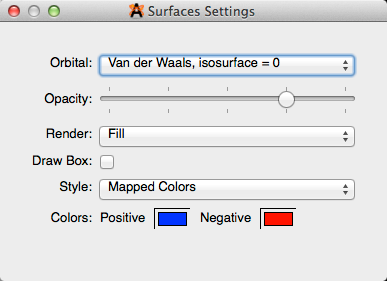
Cambiar la configuración de la superficie
La opacidad, la reproducción y los colores de la superficie se pueden cambiar haciendo clic en la llave junto al tipo de pantalla "Superficies".
Visualización de vibraciones
Esta función permite visualizar las vibraciones de un cálculo de "frecuencia" con códigos de química cuántica (por ejemplo, Gaussian, Q-Chem, etc.)
Después de abrir un archivo de salida que se ha ejecutado con la palabra clave "freq", la barra de herramientas "Vibraciones" se abrirá automáticamente.
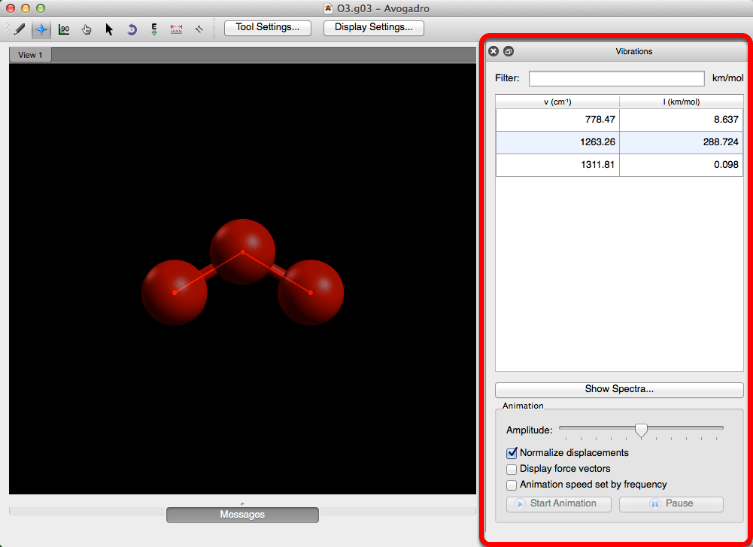
Esta barra de herramientas le permite ver las diversas vibraciones de una molécula. Seleccionando una frecuencia y haciendo clic en "Iniciar animación" comenzará una vibración.
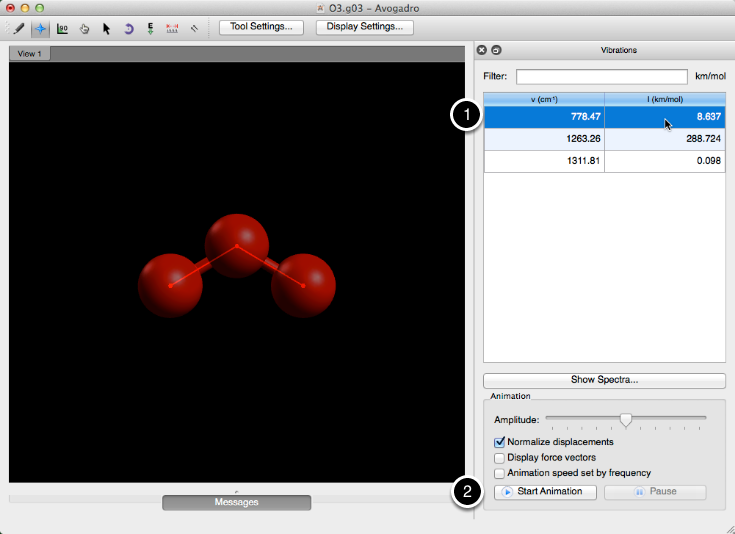
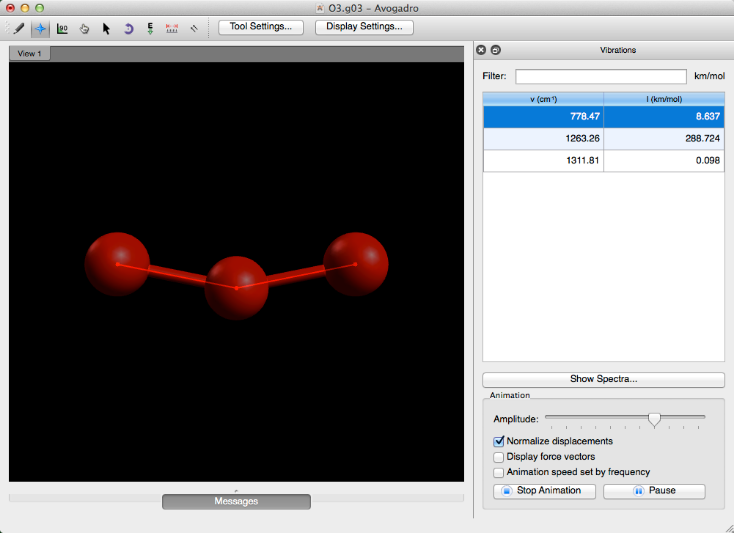
Avogadro le proporciona la capacidad de editar la amplitud de la vibración, le permite mostrar los vectores de fuerza en los átomos presentes y le permite ajustar la velocidad de la animación según la frecuencia.
Visualización de propiedades espectrales
Este tutorial cubre el trazado de espectros vibracionales, pero otros tipos de espectros son posibles con los archivos de salida de programas químicos cuánticos.
Después de abrir un archivo de salida que se ha ejecutado con la palabra clave "freq", la barra de herramientas "Vibraciones" se abrirá automáticamente.
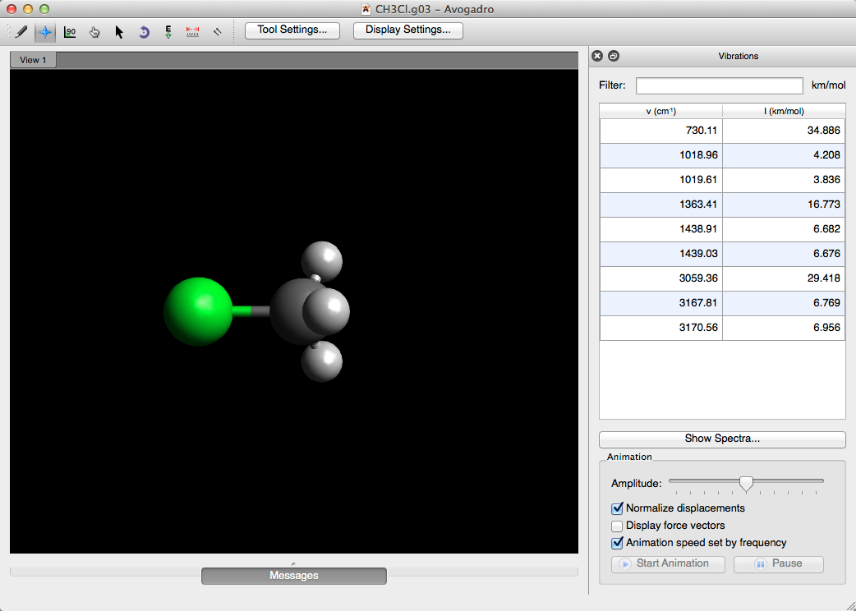
Mostrar Espectros ...
Al hacer clic en "Mostrar espectros ..." en la barra de herramientas Vibraciones, o al seleccionar "Espectros ..." en el menú "Extensiones" se abrirá una visualización de espectros de la molécula (que se muestra a continuación).
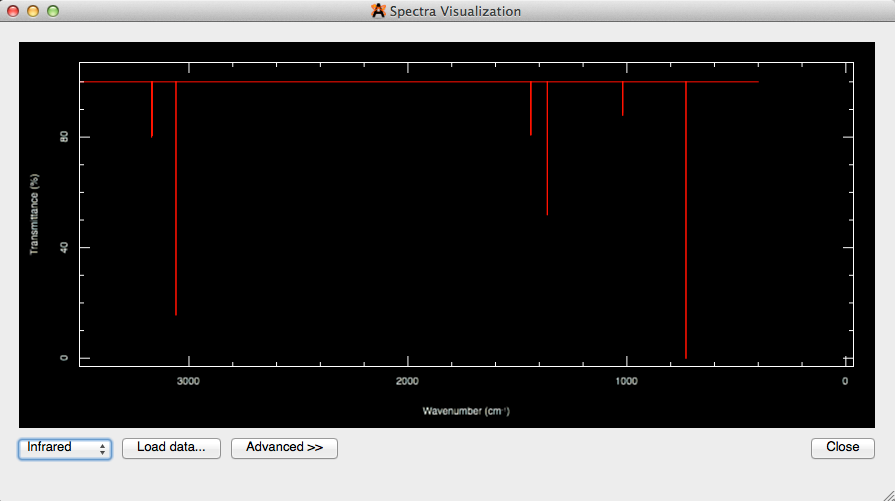
La selección de "Avanzado» en la visualización de espectros le permitirá cambiar la configuración visual, así como la configuración espectral, y exportar la imagen.
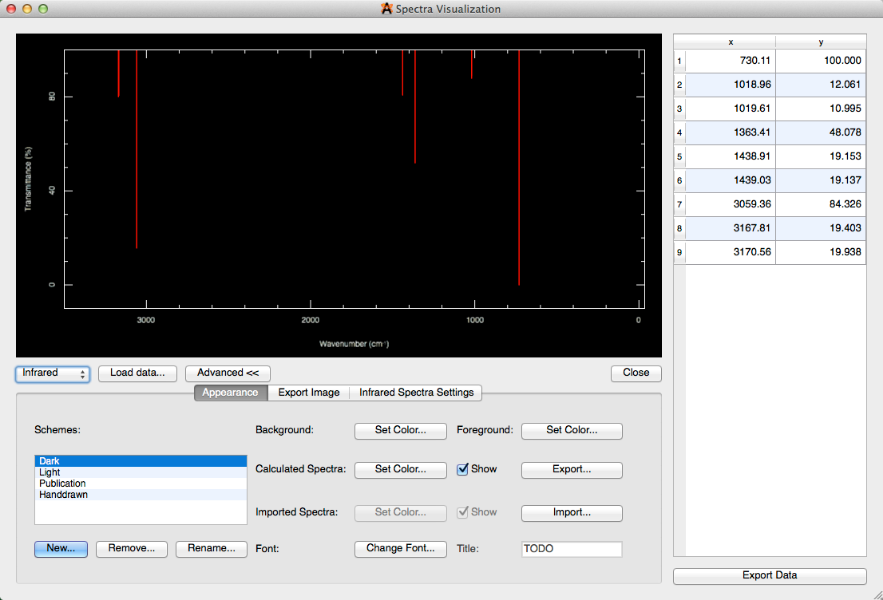
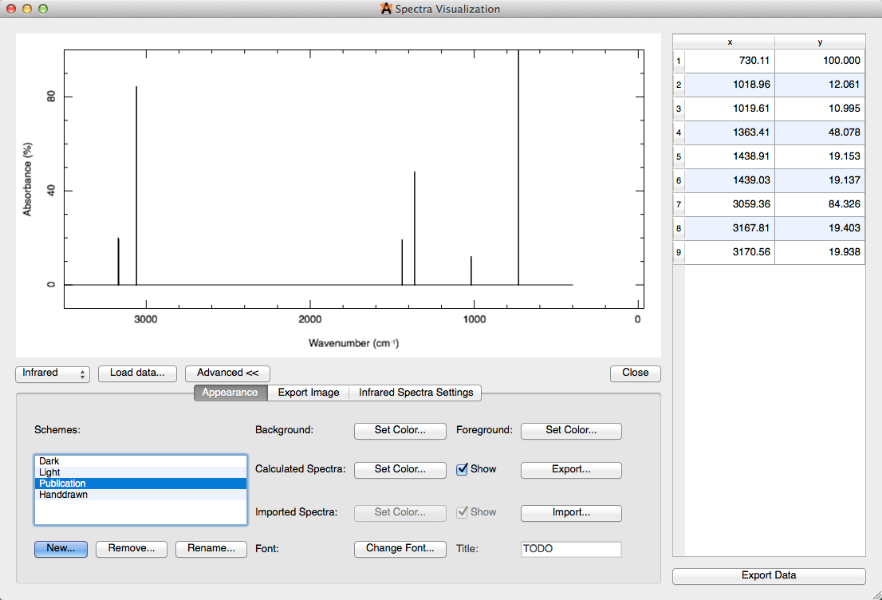
La selección "Avanzada" proporciona una multitud de ediciones generales para usar a su conveniencia. Por ejemplo, puede cambiar el% de transmitancia al% de absorbancia (en Configuración de espectros infrarrojos) y cambiar el esquema de oscuro a publicación (Configuración de aspecto).
Uso del análisis de átomos en moléculas (Bader) - QTAIM y WFN
Avogadro ahora incluye soporte para el análisis QTAIM desarrollado por el Prof. Richard Bader y su grupo. Esta técnica permite a Avogadro determinar las conexiones de enlace y las órdenes de enlace directamente desde la densidad mecánica cuántica de electrones.
Comience con un archivo WFN
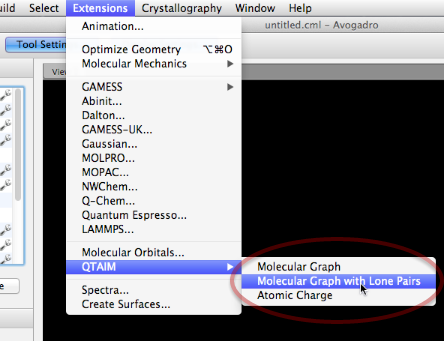
La mayoría del software de química cuántica permite crear un archivo de formato WFN a partir de la salida de un cálculo. Podemos leer este archivo a través de la extensión QTAIM para visualización y análisis. En el futuro, también se admitirán otros formatos, incluida la obtención de datos directamente desde archivos de puntos de control.
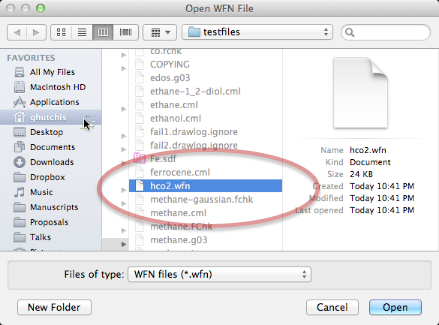
Aquí hemos seleccionado un archivo WFN de interés (HCO2) utilizando el comando Molecular Graph + Lone Pairs.
El soporte QTAIM de Avogadro leerá los átomos, determinará los enlaces y las órdenes de enlace utilizando el análisis AIM y luego continuará con el análisis adicional.
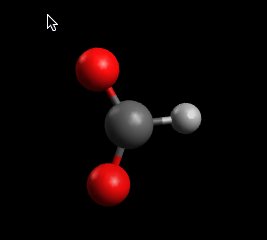
Inicialmente, puede que no veas nada diferente. QTAIM incluye un tipo de pantalla independiente para ver los resultados del análisis AIM.
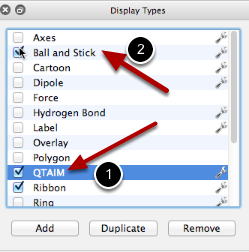
Aquí, hemos habilitado la pantalla QTAIM, que mostrará pares solitarios, puntos críticos de enlace, etc. También es posible que desee desactivar el modelo de bola y palo para ver todas las anotaciones AIM.
Aquí, también hemos desactivado el tipo clásico de visualización de bola y palo para dejar solo la vista QTAIM de HCO2.
No hay comentarios:
Publicar un comentario