Menú Archivo
El menú de archivos proporciona la capacidad estándar de crear un nuevo archivo, abrir y cerrar documentos, así como guardar documentos. También proporciona la capacidad de importar archivos de varias bases de datos.
Nuevo
La selección "Nuevo" abrirá un nuevo archivo en Avogadro.
Abierto
Después de seleccionar "Abrir", se puede acceder a un archivo que se ha guardado previamente a través del navegador emergente.
Recientemente abierto
"Abrir reciente" muestra una lista de documentos lanzados recientemente.
Cerrar
"Cerrar" cierra la ventana actualmente abierta.
Salvar
"Guardar" mantendrá su progreso.
Guardar como…
"Guardar como ..." le permite guardar el progreso sin sobrescribir el archivo original.
Volver a guardado
"Revertir a guardado" revertirá los cambios realizados en el archivo guardado anteriormente.
Importar
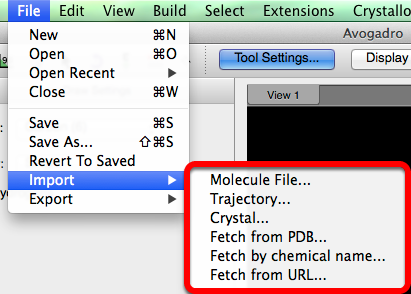
"Importar" abrirá archivos químicos almacenados en una base de datos.
Exportar
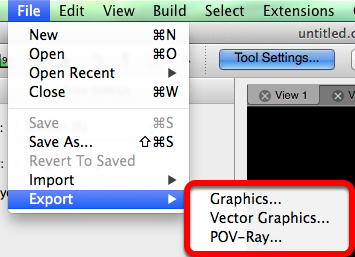
"Exportar" hará que los archivos creados en Avogadro sean adecuados para otros programas.
Menú de edición
El menú de edición administra revisiones de archivos básicas.
Deshacer
"Deshacer" anulará el último cambio al documento.
Rehacer
"Rehacer" recuperará el último cambio al documento.
Cortar
"Cortar" eliminará y copiará una selección.
Dupdo
"Copiar" creará un duplicado de la molécula completa o una selección, y lo colocará en un portapapeles.
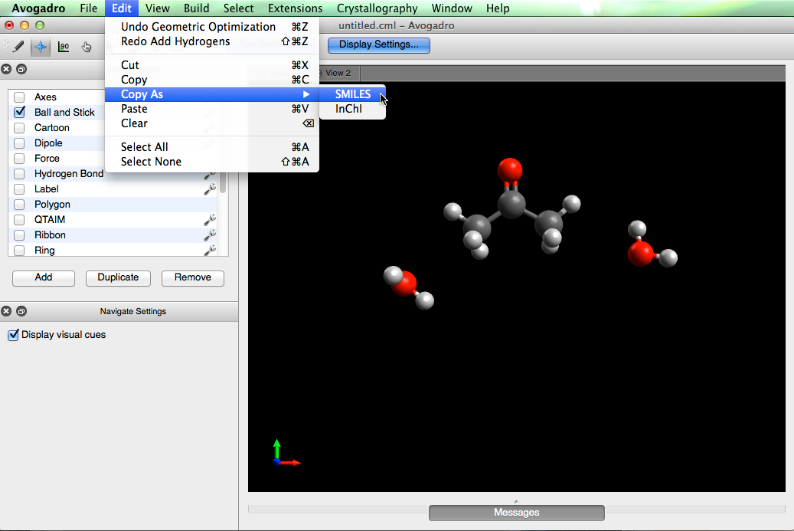
Copiar como
"Copiar como" proporciona representaciones de texto de las moléculas presentes en la pantalla de visualización. Por ejemplo, al seleccionar "Copiar como" y "SONRISAS", se muestra "C (= O) (C) COO" como salida para la pantalla de visualización a continuación. Esta selección se puede pegar en un documento de texto para proyectos externos.
Pegar
"Pegar" recuerda los últimos datos copiados en el portapapeles.
Claro
"Borrar" elimina todas las estructuras químicas de la ventana de visualización.
Seleccionar todo
"Seleccionar todo" resalta todo en la pantalla (esta función también se puede encontrar en el menú "Seleccionar").
Seleccione Ninguno
"Seleccionar ninguno" descartará todo lo que aparece en la pantalla (esta función también se encuentra en el menú "Seleccionar").
Menú ver
El menú de vista le brinda al usuario la capacidad de agregar y ajustar las vistas de visualización que se están utilizando actualmente.
Nueva vista
"Nueva vista" crea una nueva ventana de visualización en blanco.
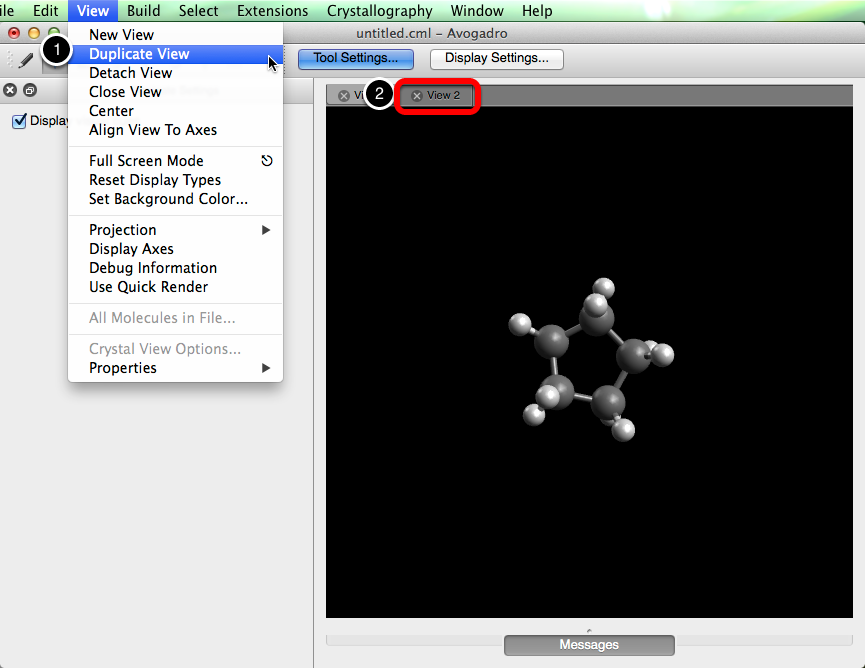
Vista duplicada
Al seleccionar "Vista duplicada" en la barra desplegable, se creará un duplicado de la vista actual. Cualquier cambio realizado en la ventana de visualización se actualizará automáticamente en todas las vistas.
Vista separada
La selección "Separar vista" mostrará la vista actual en una nueva ventana.
Cerrar vista
"Cerrar vista" elimina la pantalla que está abierta. También se puede cerrar una vista haciendo clic en la x a la izquierda de la pestaña de vista.
Centrar
"Centro" alineará la (s) molécula (s) con el centro de la pantalla de visualización.
Alinear la vista a los ejes
“Alinear vista con ejes” ajusta la vista de visualización para que esté en el plano x, y con el eje z positivo apuntando hacia usted.

Pantalla completa
"Pantalla completa" expande la ventana para llenar la pantalla de la computadora.
Restablecer tipos de pantalla
"Restablecer tipos de pantalla" anulará la selección de todos los tipos de pantalla seleccionados y volverá al tipo de pantalla predeterminado "Bola y palo".
Establecer color de fondo ...
La función "Establecer color de fondo ..." le permitirá cambiar el color de fondo de la ventana de visualización.
Proyección
Hay dos tipos de características de proyección (prospectiva y proyección ortográfica), la proyección predeterminada es la proyección en perspectiva. La proyección en perspectiva proporciona una vista más realista (3D) de una molécula en el espacio. La proyección ortográfica proporciona y ajusta la molécula en una vista plana (2D), donde todos los átomos similares se ajustan para mantener el mismo tamaño. Las vistas de proyección son más evidentes cuando se dibujan moléculas.
Ejes de visualización
“Mostrar ejes” mostrará una visualización de ejes en la esquina inferior izquierda.
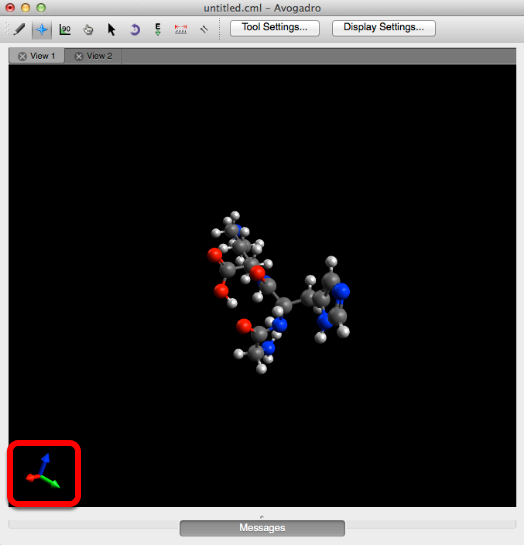
Información de depuración
La “Información de depuración” proporciona información adicional sobre la vista y lo que está ocurriendo actualmente en su pantalla.
Usar render rápido
El procesamiento rápido ajusta la imagen molecular 3D en la pantalla de visualización para lograr un procesamiento más rápido de la imagen en computadoras lentas. En la mayoría de las computadoras modernas (2012 o posteriores), esto no es necesario.
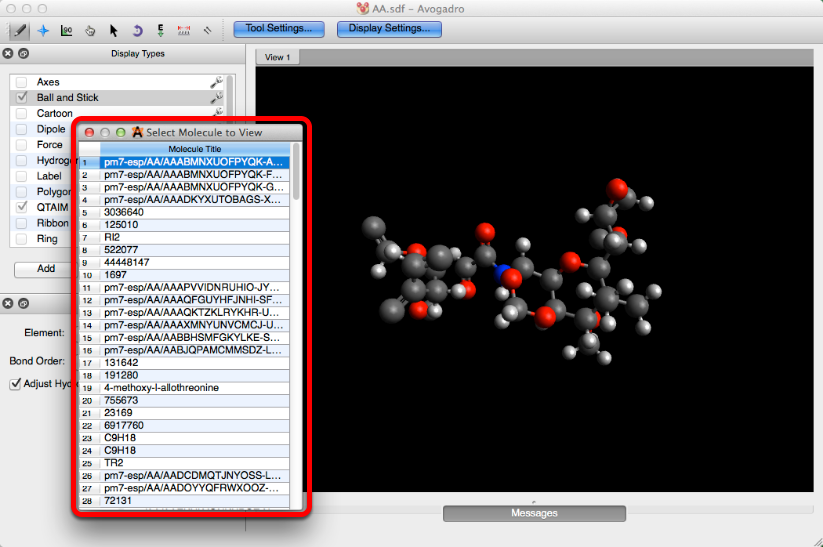
Todas las moléculas en archivo ...
"Todas las moléculas en el archivo ..." le permite ver todas las moléculas que se crearon e integraron previamente en un archivo. Desde el cuadro de diálogo que aparece, puede seleccionar y editar una molécula haciendo clic en el título de la molécula.
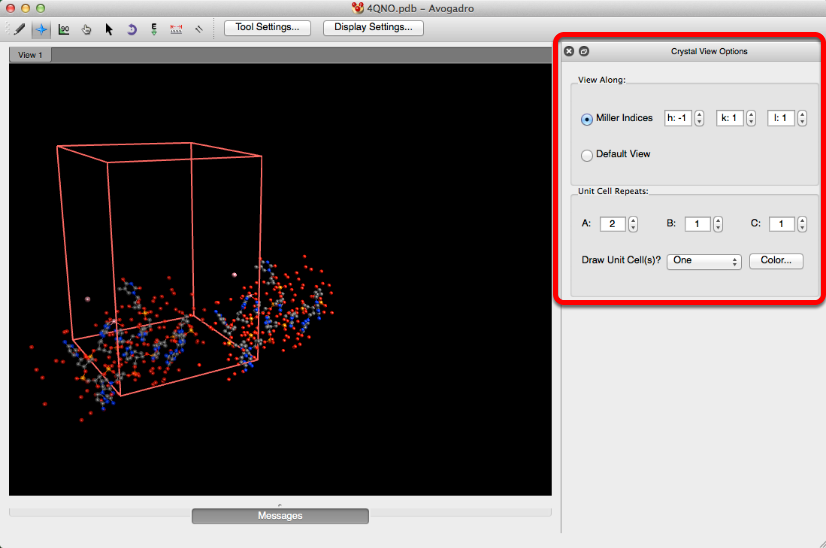
Opciones de vista de cristal ...
"Opciones de vista de cristal ..." cuando se seleccione, abrirá la barra de herramientas que se muestra a continuación. Esta barra de herramientas le permite editar los índices de Miller y la celda unitaria para cualquier estructura de cristal.
Propiedades
La selección de "Propiedades" le proporcionará las propiedades de molécula, átomo, enlace, ángulo, torsión y conformador. Estas configuraciones muestran información general de composición sobre la molécula y los átomos presentes.
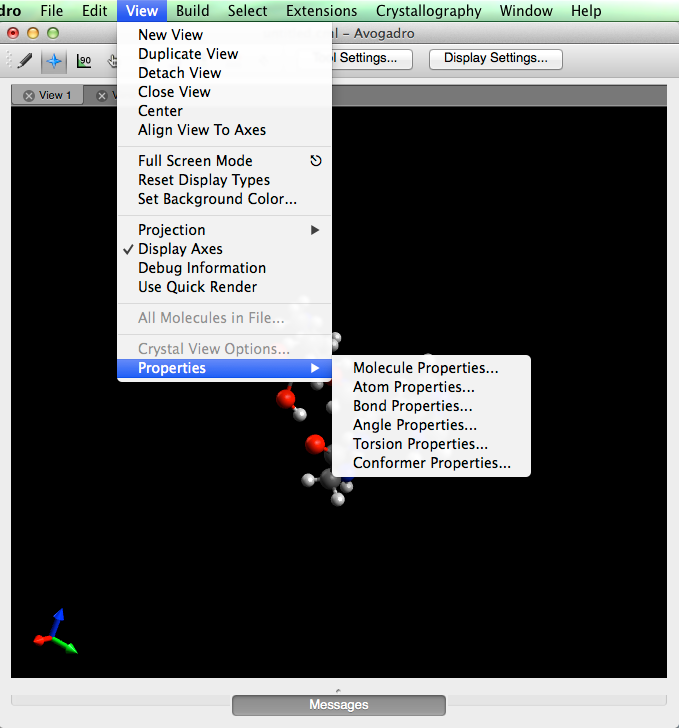
Por ejemplo, al hacer clic en "Propiedades de la molécula" se mostrará información molecular general.
Menú de compilación
El menú de construcción ayuda a facilitar el proceso de construcción de moléculas.
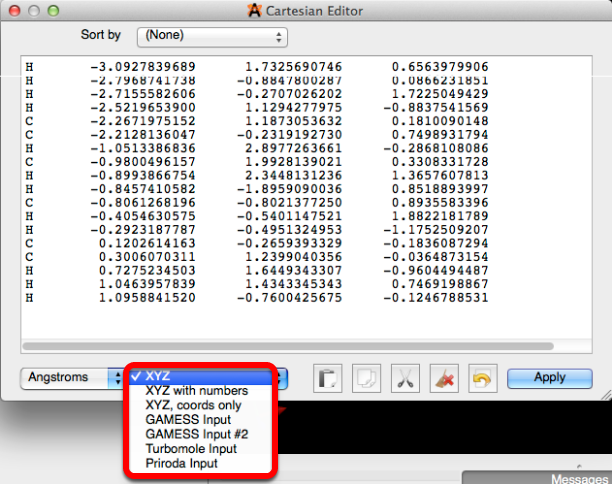
Editor cartesiano…
El "Editor cartesiano ..." cuando se selecciona le proporciona la capacidad de ajustar manualmente las longitudes de unión. El cuadro de diálogo para el editor cartesiano se muestra a continuación.
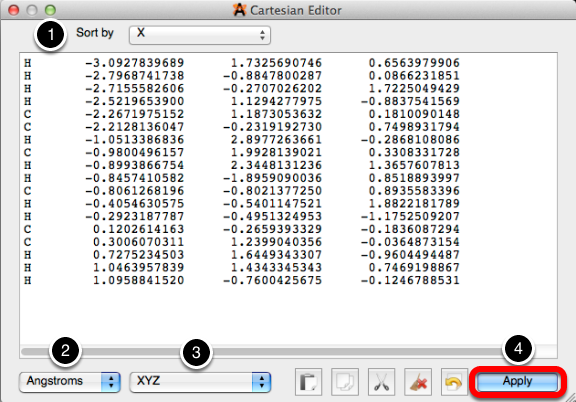
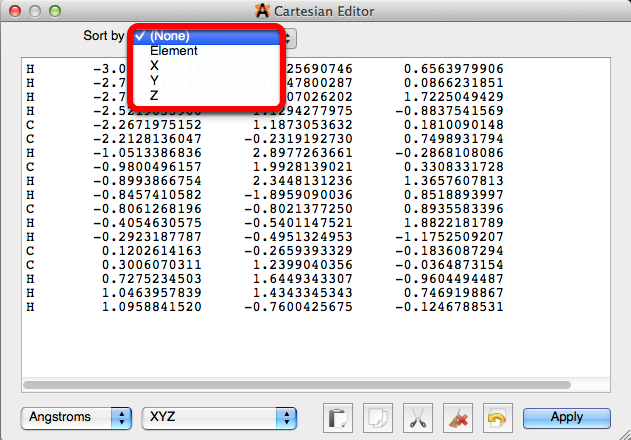
1. Ordenar por ...
El menú desplegable de ordenación reorganizará los datos en el cuadro de diálogo para su conveniencia. Ordenar por puede organizar los datos por elemento, o por ubicación del átomo. Todos los datos para clasificar por coordenadas X, Y y Z comenzarán a leer en el átomo hasta el extremo más alejado en la molécula, y continuarán hasta que alcancen el átomo en el punto más a la derecha.
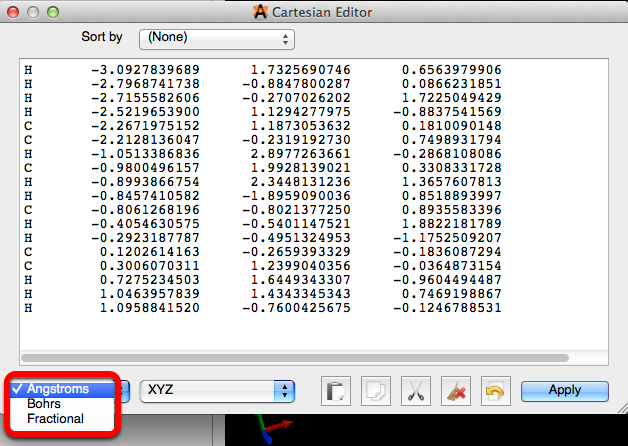
2. Unidad de medida
Avogadro proporciona tres unidades de medida para ajustar longitudes de enlace, Angstroms, Bohrs y coordenadas fraccionales. Una celda unitaria debe estar definida para usar coordenadas fraccionarias.
3. Edición y modificación de datos
Editar los datos es tan simple como hacer clic en el número que desea editar y escribir una nueva coordenada. Después de hacer clic en "Aplicar" y volver a la pantalla de visualización de Avogadro, debe notar que el átomo ha cambiado de posición.
Los datos mostrados también se pueden modificar de acuerdo con sus preferencias personales, o para el uso de complementos adicionales.
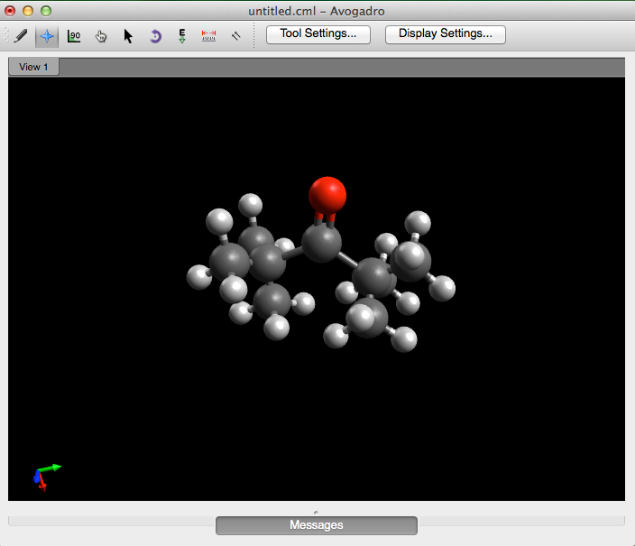
Cambia H a Metil
"Cambiar H a metilo" reemplazará cualquier hidrógeno presente en la ventana de visualización con grupos metilo. Abajo se muestra la acetona con todos sus hidrógenos reemplazados por grupos metilo.
Añadir hidrógenos
"Agregar hidrógenos" satisfará la valencia de los átomos presentes con los hidrógenos.
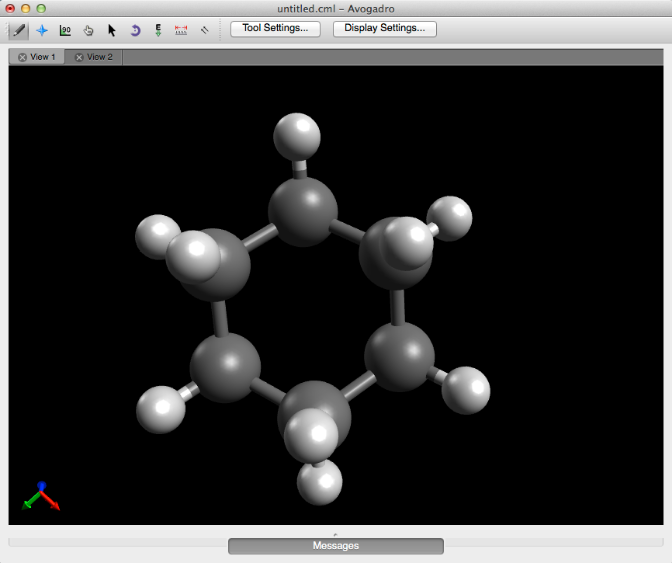
Añadir hidrógenos para pH ...
"Agregar hidrógenos para pH ..." creará un cuadro de diálogo (que se muestra a continuación) que le permite ajustar el pH del entorno molelcular. Esta característica agregará (o restará) hidrógenos ácidos a grupos ionizables en péptidos, de acuerdo con el pH deseado.
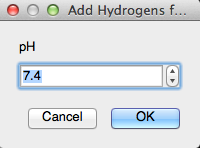
Eliminar hidrógenos
"Eliminar Hidrógenos" eliminará todos los hidrógenos en la pantalla de visualización.
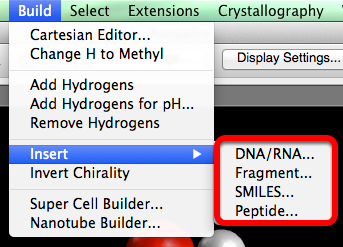
Insertar
"Insertar" proporciona una forma más rápida y sencilla de construir moléculas. Como se muestra a continuación, puede insertar ADN / ARN, un fragmento, un péptido y también puede insertar una molécula basada en el texto SONRISAS.
Invertir la quiralidad
"Invertir quiralidad" invertirá (invertirá) la quiralidad inicial a la configuración R / S opuesta.
Super Cell Builder ...
La información sobre Super Cell Builder se puede encontrar en la sección "Construcción y edición de cristales y materiales".
Constructor de nanotubos ...
La información sobre la construcción de nanotubos de carbono en Avogadro se puede encontrar en la sección "Moléculas de construcción".
Menú Select
El menú de selección hace que las alteraciones químicas sean más eficientes a través de varios modos de selección.
Seleccionar todo
"Seleccionar todo" resalta todo lo que aparece en la pantalla (esta función también se puede encontrar en el menú "Editar").
Seleccione Ninguno
"Seleccionar ninguno" descartará todo lo que aparece en la pantalla (esta función también se encuentra en el menú "Editar").
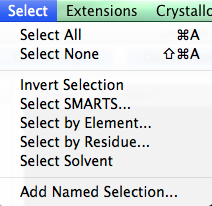
Invertir selección
"Invertir selección" invierte la selección realizada.
La primera imagen que se muestra a continuación es la selección original, y la segunda muestra la selección invertida.
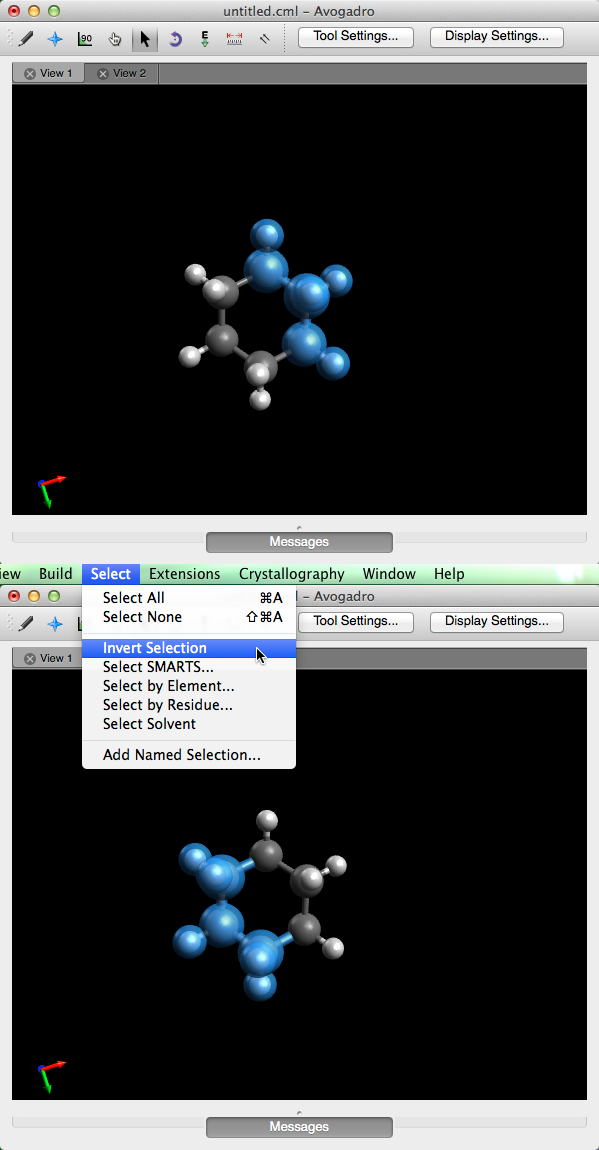
Seleccione SMARTS ...
SMARTS ( SMiles ARbitrary Target Specification) es una extensión del lenguaje químico más general de SMILES. "Seleccionar SMARTS ..." le permite usar este lenguaje químico para seleccionar varios átomos o grupos de átomos dentro de la molécula. Por ejemplo, si escribe "a" en el cuadro de diálogo y hace clic en Aceptar, seleccionará todos los átomos con aromaticidad. Puede encontrar más información en la página web de Daylight SMARTS .
Seleccionar por elemento ...
"Seleccionar por elemento ..." genera una pantalla emergente de tabla periódica que le permite seleccionar un elemento en toda la pantalla de visualización.
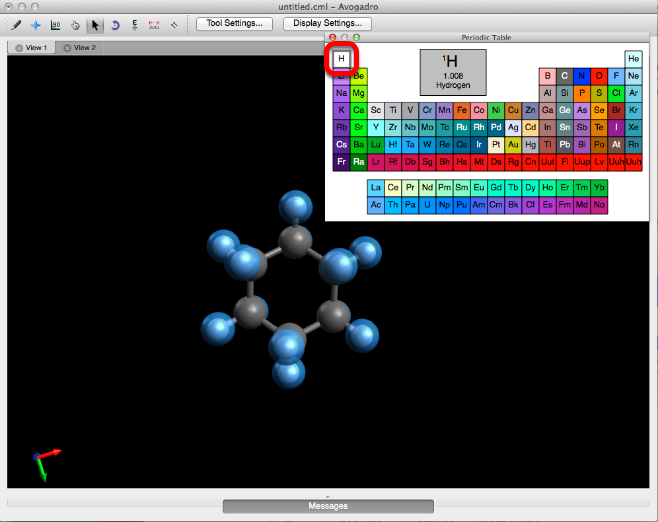
Seleccionar por Residuo ...
"Seleccionar por residuo ..." genera una pantalla emergente que le permite seleccionar residuos con nombres específicos. Por ejemplo, al escribir "ALA" (esta función distingue entre mayúsculas y minúsculas) para seleccionar un residuo de alanina.
Esta característica solo funciona con residuos que se crearon utilizando el generador de péptidos.
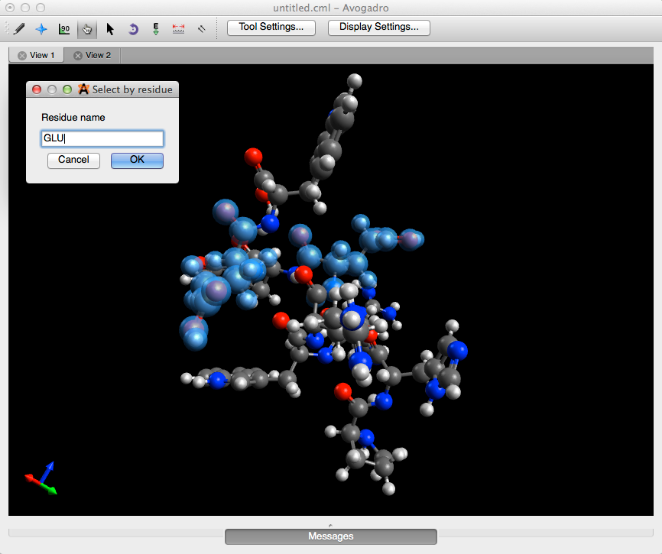
Seleccionar solvente
Esta función seleccionará residuos "HOH" en archivos PDB (banco de datos de proteínas).
Añadir selección de nombre ...
"Agregar selección de nombre ..." le permite agregar una nueva selección a la base de datos actual, que luego puede recordar a su conveniencia.
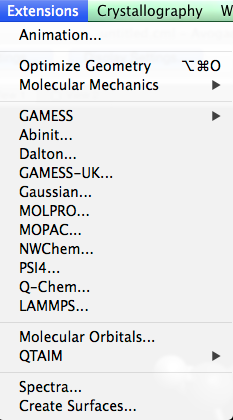
Menú de extensiones
El menú Extensiones es un catálogo de complementos computacionales equipados con Avogadro. Estos complementos pueden interactuar con moléculas, generar diálogos de archivo de entrada para códigos cuánticos y crear diálogos de propiedades de moléculas.
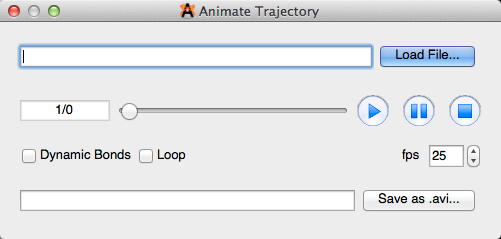
Animación
Al seleccionar "Animación" se abrirá el cuadro de diálogo de la trayectoria animada que se muestra a continuación. Desde aquí puede cargar un archivo, ver y editar la animación, así como guardar el archivo en un formato compatible con PC.
Optimizar la geometría
"Optimizar Geometría" proporciona una representación rápida y realista de una molécula utilizando la mecánica molecular.
Mecánica Molecular
"Mecánica molecular" le permite editar la optimización de geometría de una molécula, para que se adapte mejor a sus propósitos.
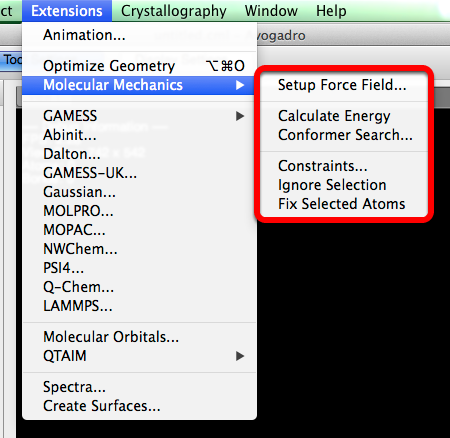
Configurar campo de fuerza ...
Se abrirá un cuadro de diálogo cuando se seleccione "Configurar campo de fuerza ...". Este cuadro de diálogo le brinda la posibilidad de elegir el tipo de campo de fuerza y el algoritmo que mejor optimice sus parámetros moleculares y preferencias.
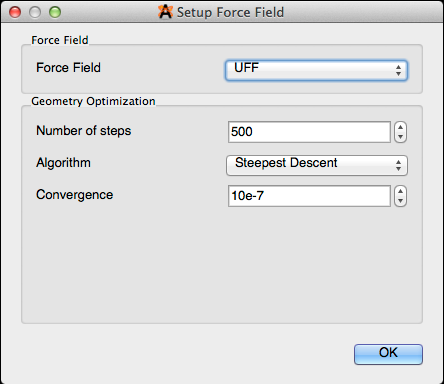
Calcular energía
"Calcular energía" determina la cantidad de energía por la cantidad de material (kJ / mol) y muestra este número en un cuadro de diálogo emergente.
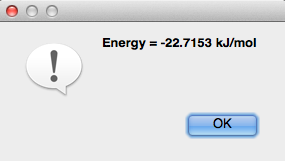
Búsqueda de conformadores
La "Búsqueda de Conformador" es una forma de buscar fácilmente a los conformadores dentro de una molécula (el cuadro de diálogo se muestra a continuación). En la sección "Optimización de la geometría" de este manual se encuentra un resumen más detallado sobre cómo realizar una búsqueda de conformidad. Avogadro solo rinde conformaciones escalonadas y no calcula conformadores de anillos.
Restricciones
"Restricciones" es una forma de garantizar la estabilidad del átomo en varias selecciones (cuadro de diálogo que se muestra a continuación). Las restricciones que se pueden aplicar a una molécula incluyen Ignorar el átomo, Arreglar el átomo, Arreglar el átomo X, Arreglar el átomo Y, Arreglar el átomo Z, Distancia, Ángulo y Ángulo de torsión. En la sección "Optimización de la geometría" de este manual se encuentra un resumen detallado de cómo usar la función de restricciones.
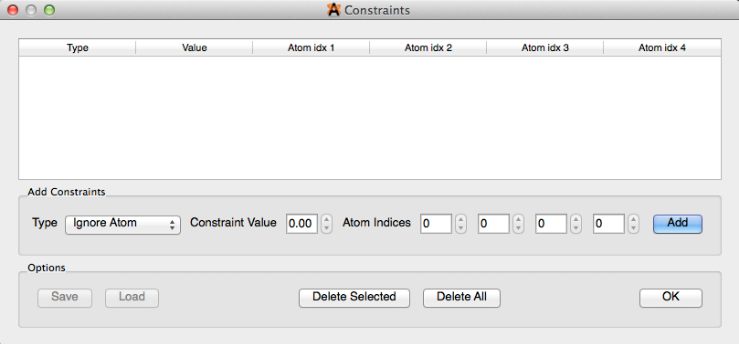
Ignorar la selección
"Ignorar selección" le permite seleccionar una parte específica de una molécula para omitirla durante una optimización de geometría.
Arreglar los átomos seleccionados
“Arreglar los átomos seleccionados” también le permite configurar una cierta parte de una molécula para corregirla durante la optimización.
Extensiones de Avogadro – Plugins
Avogadro le proporciona la capacidad de conectar sus moléculas con otros complementos basados en diálogo. Estas extensiones interactúan con una molécula para proporcionar más información molecular y capacidades de cálculo adicionales. Estos complementos incluyen, entre otros, GAMESS, Abinit, Dalton, GAMESS-UK, Gaussian, MOLPRO, MOPAC, NWChem, PSI4, Q-Chem y LAMMPS.
General "Cómo" para los complementos
Avogadro (como se verá más adelante) se puede utilizar para mostrar orbitales moleculares, QTAIM, espectros, así como para crear superficies. Sin embargo, muchas de estas funciones no se pueden utilizar en todo su potencial sin ejecutar primero uno de los complementos enumerados en la sección anterior. Gaussian es uno de los complementos más utilizados, debido a su amplia gama de conjuntos / funciones básicas.
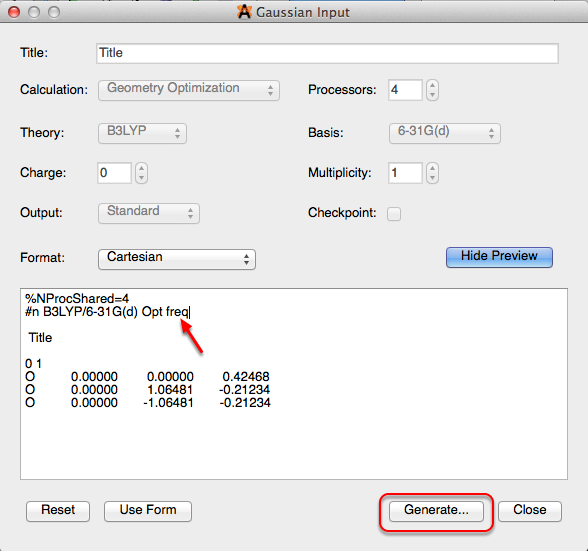
Corriendo gaussiano
Después de seleccionar "Gaussian" en el menú de Extensiones, aparecerá el cuadro de diálogo que se muestra a continuación. Puede editar el cuadro de diálogo y agregar palabras clave específicas para utilizar estas funciones en Avogadro. Por ejemplo, al escribir "freq" en el cuadro de diálogo se calcularán las constantes de fuerza y las frecuencias vibratorias. Puede encontrar más información sobre palabras clave para gaussiano en el sitio web de Gauss (http://www.gaussian.com/g_tech/g_ur/l_keywords09.htm). Luego, hacer clic en Generar le permitirá guardar el archivo en su computadora, para que pueda ejecutar el archivo en un software externo.
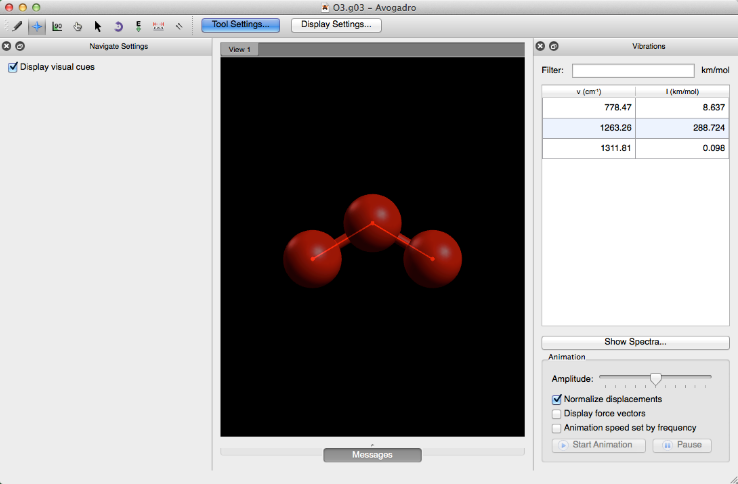
Una vez que el archivo se haya ejecutado a través del software externo, tendrá un archivo .g03 o .g09 que abrirá la selección de palabras clave en una barra de herramientas en el lado derecho de la pantalla. "Freq" abrirá la barra de herramientas de vibraciones que se muestra a continuación.
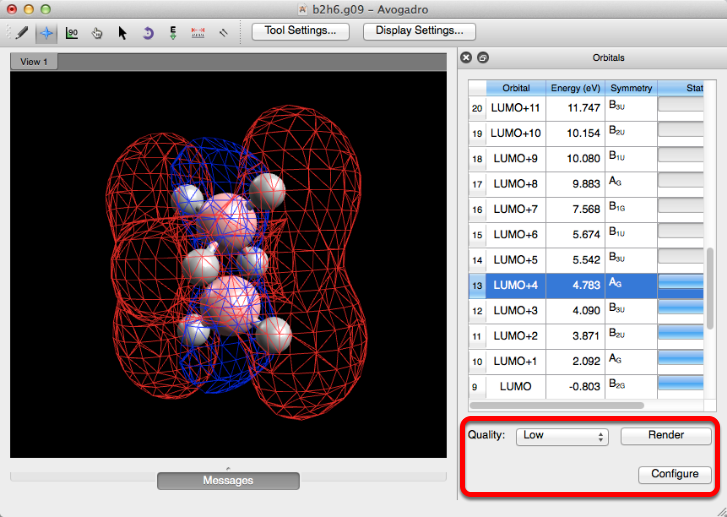
Orbitales moleculares
La selección de "Orbitales moleculares" mostrará los orbitales moleculares para los orbitales con barras de estado completas. La calidad de los orbitales se puede ajustar y reconfigurar si es necesario. Esta función solo funciona ejecutando archivos de extensión gaussianos (.fchk, .g03, .g09, etc.).
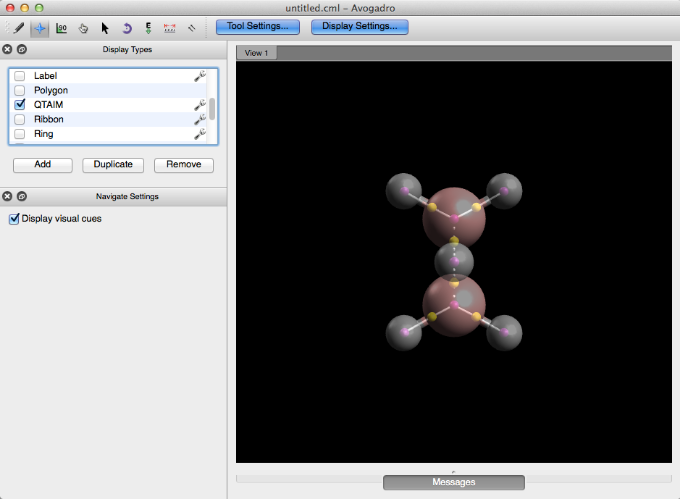
QTAIM (Teoría Cuántica de Átomos en Moléculas)
QTAIM muestra el enlace implícito que se teoriza para tener lugar entre los hidrógenos de los cristales orgánicos (el enlace implícito se transmite a través de puntos). Este tipo de pantalla se utiliza importando un archivo .wfn de la selección "QTAIM", "Molecular Graph" en el menú "Extensions". La selección de "Gráfico molecular con pares solitarios" o "Carga atómica" proporcionará información concurrente sobre la molécula. Puede encontrar más información sobre este proceso en la sección Tutorial de este manual.
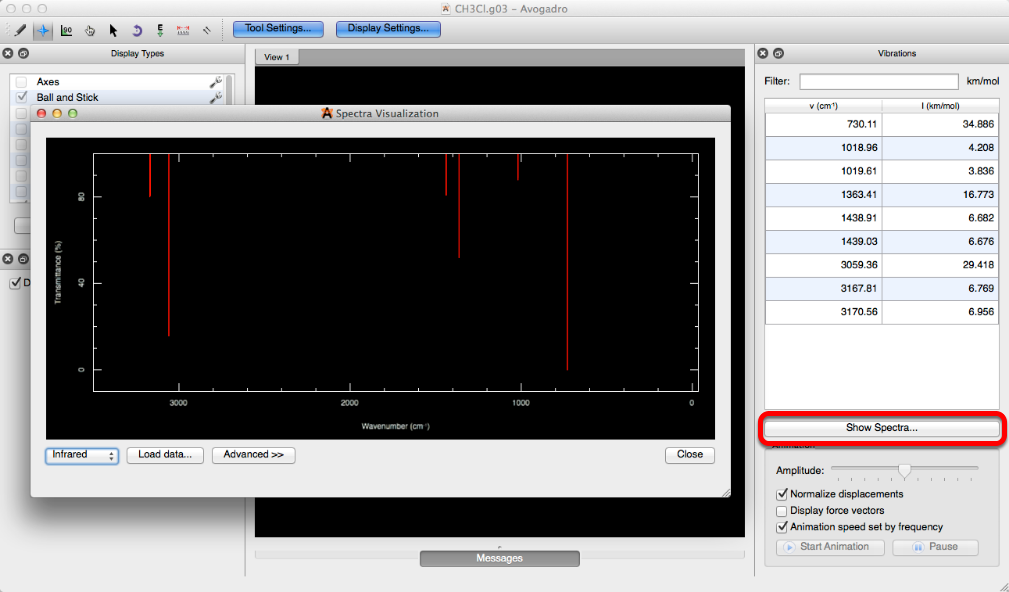
Espectros ...
Al hacer clic en "Espectros ..." se creará una visualización de espectros de un archivo .g03 o .g09 que se ha ejecutado con la palabra clave "freq". También se puede crear una visualización espectral a través de la barra de herramientas de vibraciones seleccionando "Mostrar espectros ...".
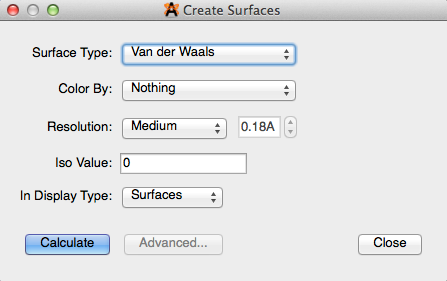
Crear superficies ...
"Crear superficies ..." le permite ver las superficies de Van der Waals, potencial electrostático, densidad electrónica y superficies orbitales moleculares. Las opciones de tipo de superficie para la visualización dependen de qué tipo de cálculos se han ejecutado previamente en la molécula. El tipo de archivo que abre / crea permite más o menos opciones de visualización en la superficie (generalmente discutidas en Avogadro Extensions – Plugins). Esta función también le permite editar el color, la resolución y el valor iso para mejorar aún más su superficie.
No hay comentarios:
Publicar un comentario