Herramienta de dibujo
1. Creando una Molécula
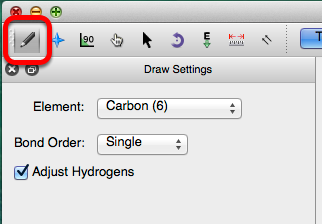
Las moléculas se pueden construir y editar con la herramienta de dibujo.
2.
Puedes comenzar a crear una molécula haciendo clic izquierdo en la pantalla negra. Esto generará un átomo de carbono. Haciendo clic derecho en el átomo lo borrará.
3.
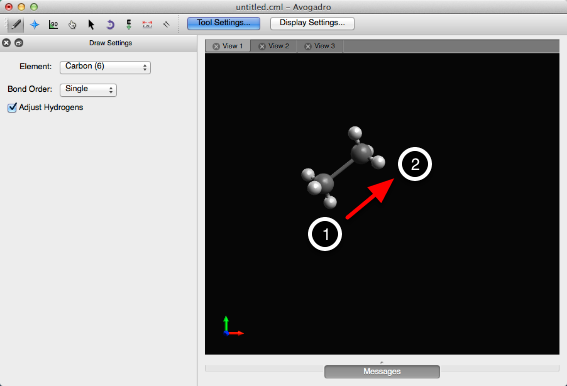
Si hace clic con el botón izquierdo y arrastra el mouse, se generará un vínculo con otro átomo de carbono.
4.
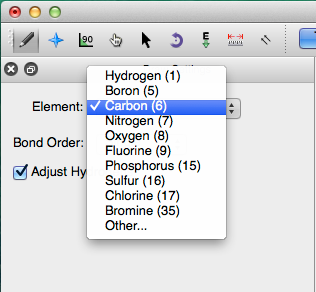
Avogadro utiliza el carbono como elemento predeterminado. Se puede seleccionar un elemento diferente a través del menú desplegable "Elemento". Escribir el símbolo atómico (por ejemplo, "Como" para Arsénico) es un atajo para cambiar el elemento seleccionado.
5.
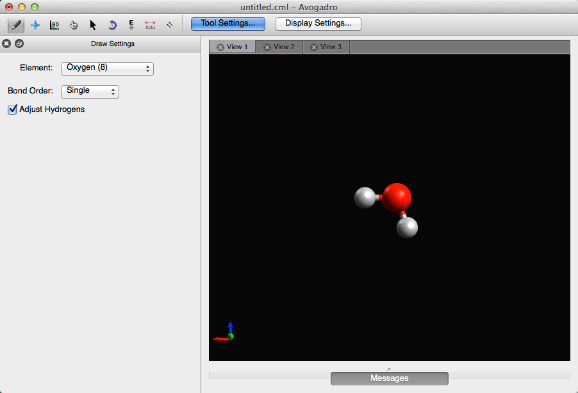
Digamos que querías crear agua. Puede escribir "O" o seleccionar "Oxygen (8)" en el menú desplegable y luego hacer clic en la pantalla negra.
Al hacer clic con el botón izquierdo en un átomo que ya se ha generado también se cambiará el elemento. En este caso, hacer clic en el átomo de carbono inicial lo convirtió en un átomo de oxígeno.
6.
Si se marca la casilla "Ajustar hidrógenos", los átomos de hidrógeno en la molécula se ajustan automáticamente para satisfacer la valencia (como se muestra arriba).
7.
La orden de los bonos se cambia a través del menú desplegable "Orden de los bonos" o escribiendo los números "1", "2" o "3". Los bonos se agregan haciendo clic izquierdo en un enlace que ya ha sido creado. Al hacer clic con el botón derecho en un enlace, se elimina el enlace y el átomo al que está vinculado.
8.
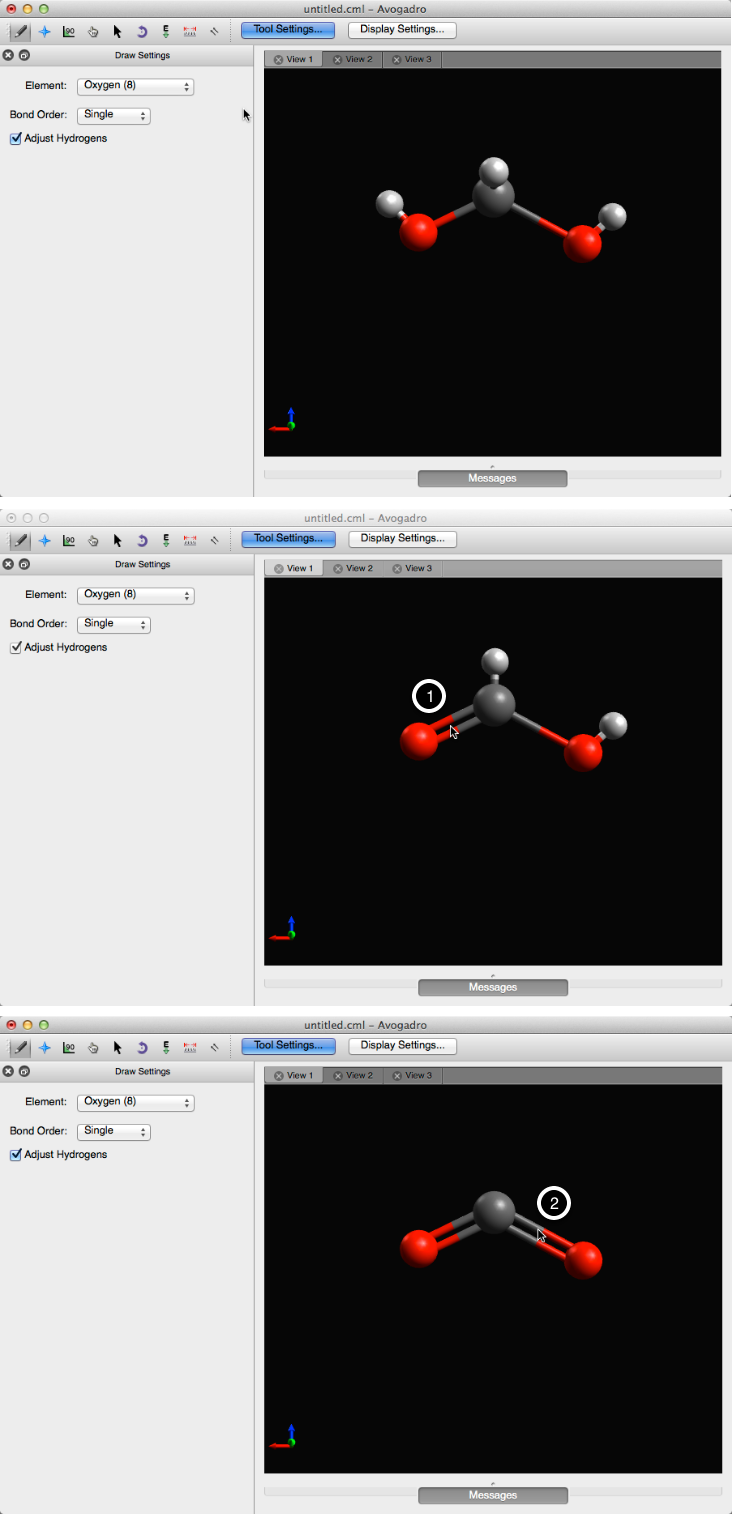
Creación de dióxido de carbono:
Comienza a dibujar la estructura “OCO”. Después de dibujar la estructura, todo lo que necesita hacer es hacer clic izquierdo en los enlaces. Al hacer clic con el botón izquierdo en los enlaces se creará un enlace doble (como se muestra a continuación).
9.
Una vez que haya creado su molécula, puede optimizar su geometría a través del menú de extensiones. Seleccionar el menú "Extensiones" y hacer clic en "Optimizar geometría" proporcionará a su molécula las longitudes y ángulos de enlace adecuados.
Herramienta de navegación
La herramienta de navegación se utiliza para desplazar, rotar y escalar la vista de una molécula.
Si la casilla "Mostrar indicaciones visuales" está marcada, las flechas amarillas mostrarán qué tipo de navegación está teniendo lugar.
Al hacer clic en cualquier lugar de la pantalla negra y arrastrar el mouse, se inclinará y rotará la molécula completa (como se muestra a continuación).
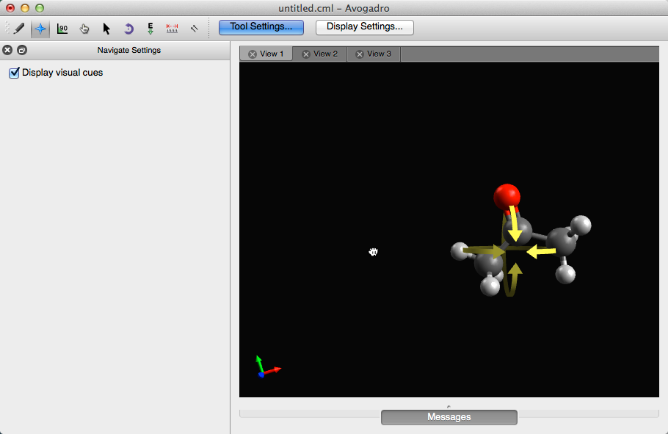
Una molécula también puede rotarse sobre un átomo haciendo clic en el átomo y arrastrando el mouse. Representado a continuación, la acetona se gira alrededor de su carbono inicial.
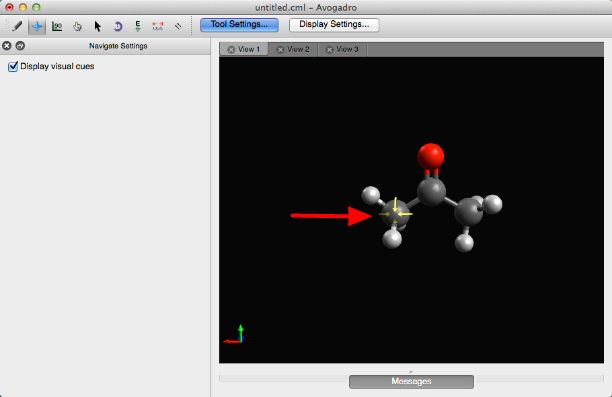
El clic derecho le permite cambiar la ubicación de las moléculas en la pantalla. Al hacer doble clic en la molécula se restablecerá la vista de la molécula.
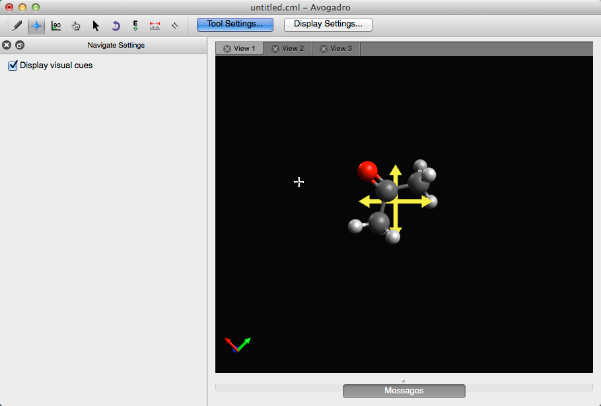
El uso de la barra de desplazamiento central de un mouse le permitirá acercar y alejar la imagen.
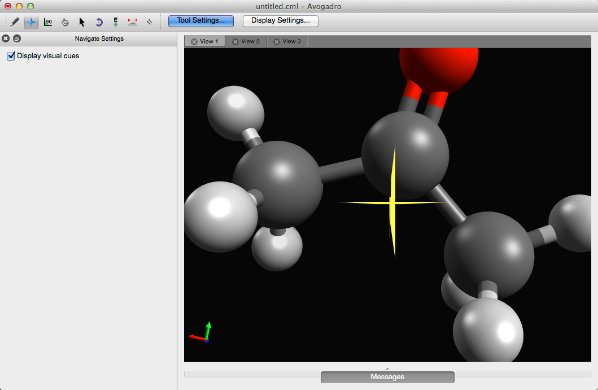
Herramienta de manipulación de Bond Centric
La herramienta de manipulación centrada en enlaces modifica los ángulos, enlaces y torsiones de una molécula.
#### Comenzando con lo básico:
- Para comenzar a utilizar la herramienta de manipulación centrada en el enlace, haga clic en un enlace dentro de su molécula.
- Haciendo clic en un enlace y arrastrando el cursor le permite ajustar el plano.
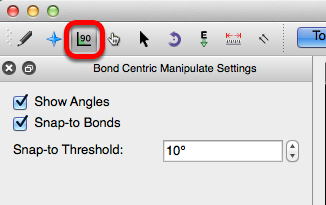
- Si se marca la casilla "Mostrar ángulos", se muestran los ángulos del enlace seleccionado a todos los enlaces adyacentes.
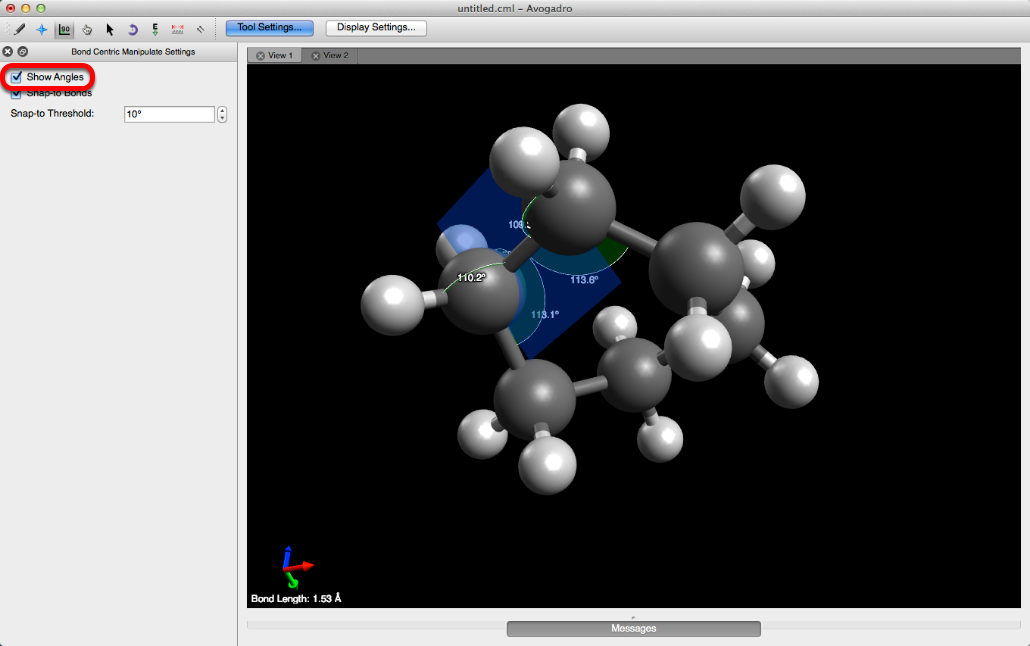
- Si se marca la casilla "Snap-to Bonds" notará que el plano cambia de amarillo a azul a medida que se gira. Un plano amarillo indica que un enlace adyacente está en línea con el plano. Si el plano representado es azul, el plano no estáalineado con ningún enlace adyacente.
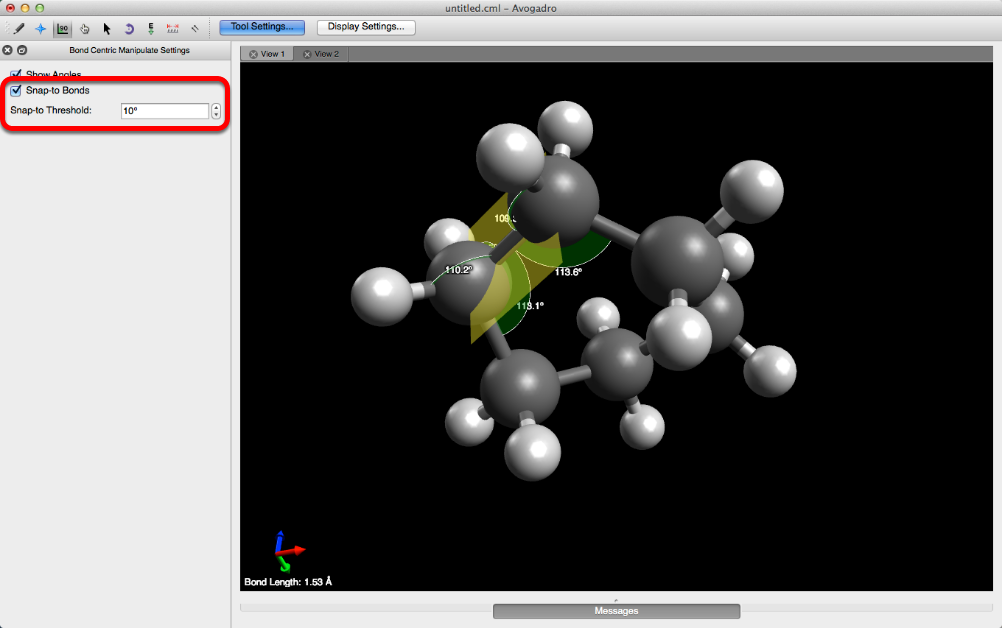
- “Snap-a Threshold” determina cuántos grados de distancia de un avión tiene que ser a ajustar a un plano de la unión adyacente.
- Por ejemplo, el "Umbral de ajuste" que se muestra a continuación es de 10 grados. Por lo tanto, si el plano que se gira gira dentro de los 10 grados de un enlace adyacente, se ajustará al plano del enlace adyacente.
- Cambiar el umbral de ajuste a 90 grados es un truco rápido para que el plano giratorio se ajuste solo a los planos de enlace adyacentes.
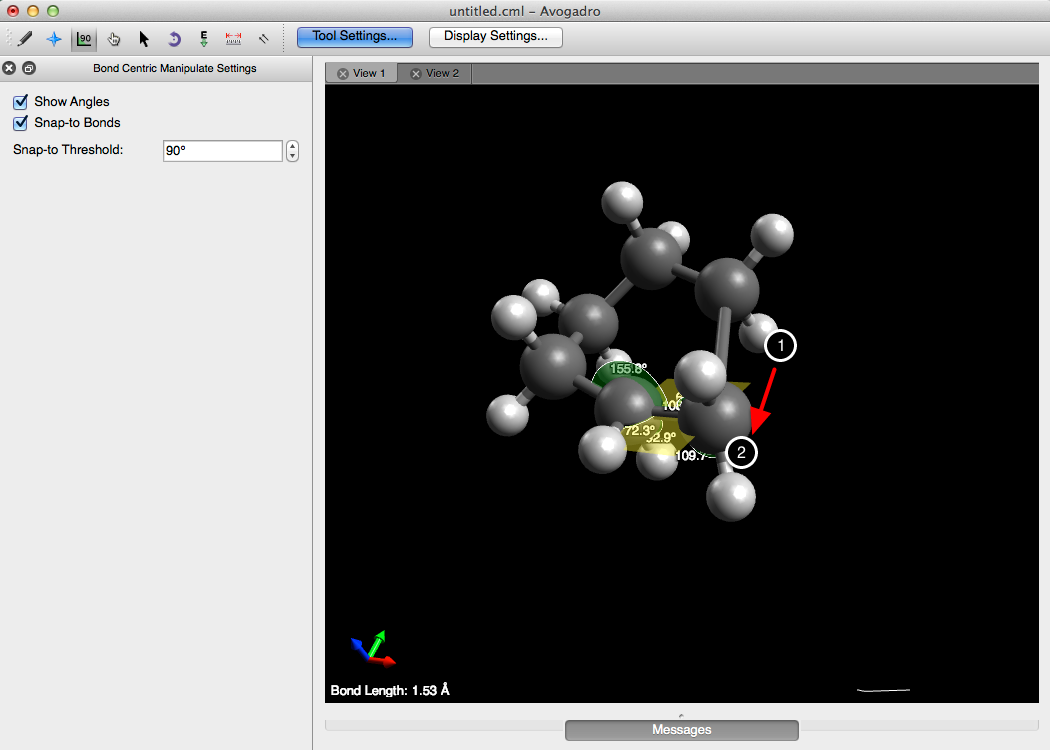
Ajuste de enlaces y ángulos de torsión
Una vez que se selecciona un plano, los átomos en cada extremo del plano pueden ser manipulados, haciendo clic izquierdo en el átomo y arrastrando. Los ángulos mostrados se ajustarán automáticamente, y el enlace seleccionado nocambiará en longitud.
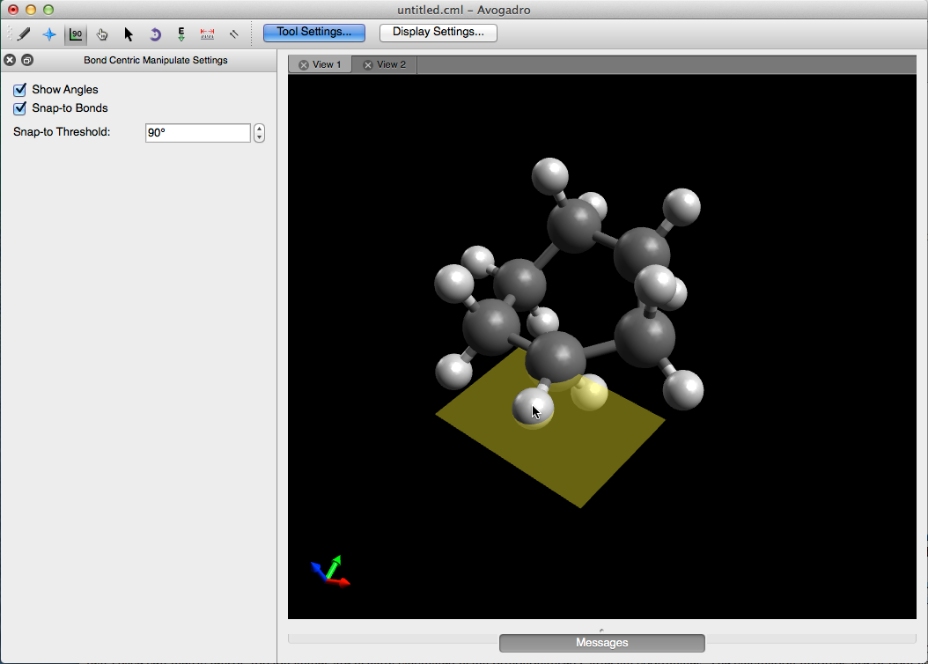
Haga clic con el botón izquierdo en un sustituyente o, en este caso, un enlace de hidrógeno a uno de los átomos de carbono seleccionados le permitirá ajustar el ángulo de torsión.
Herramienta de manipulación
La herramienta de manipulación le permite mover átomos y fragmentos seleccionados.
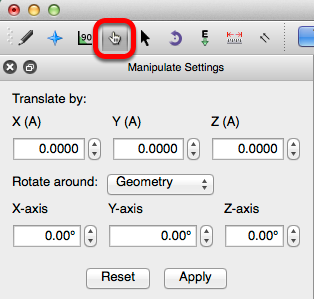
La herramienta de manipulación se selecciona automáticamente al insertar un fragmento. Esto permite que el fragmento se gire a su posición.
La sección "Traducir por:" le permite traducir fácilmente la longitud de un enlace (a un fragmento o átomo) a la posición deseada. Para que se realice una traducción, ingrese la traducción deseada y luego haga clic en "Aplicar".
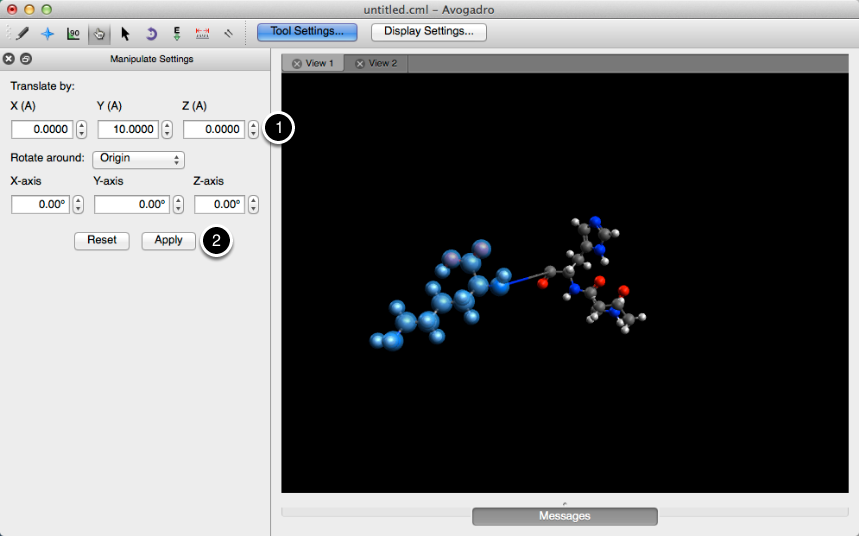
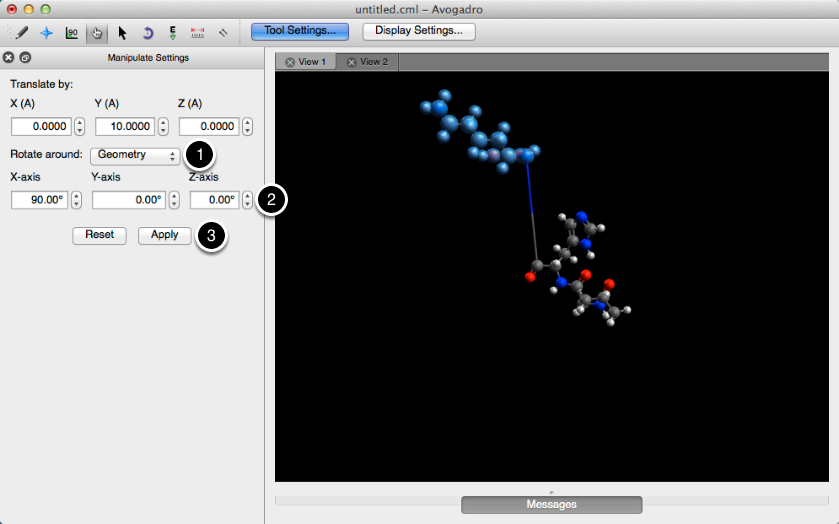
La sección "Girar alrededor de:" le permite rotar su selección alrededor de la geometría actual, o el origen. Después de elegir cuántos grados para rotar su selección, haga clic en "Aplicar".
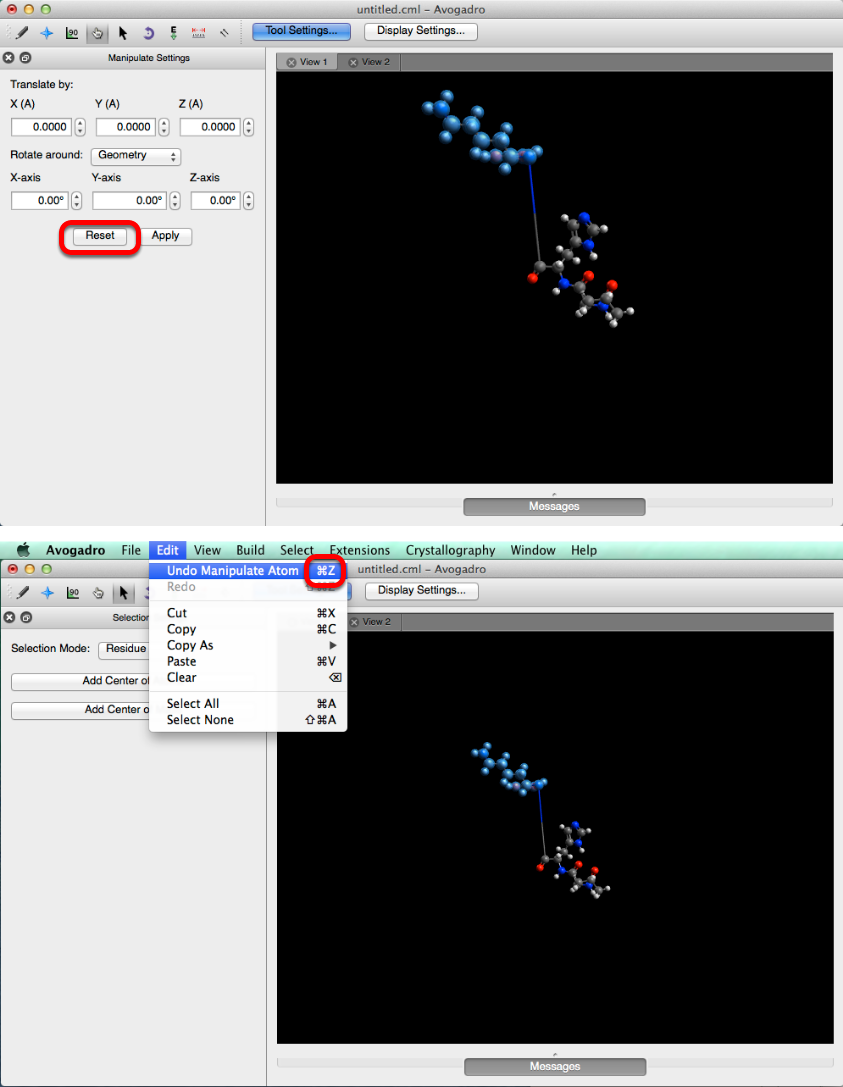
Al hacer clic en "Restablecer" se restablecerá toda la información en los cuadros de traducción y rotación. Esto no restablece la molécula. Si desea deshacer sus ajustes, vaya al menú "Editar" en la barra superior y seleccione "Deshacer Manipular Átomo".
Herramienta de selección
La herramienta de selección permite la selección individual de átomos, enlaces o fragmentos.
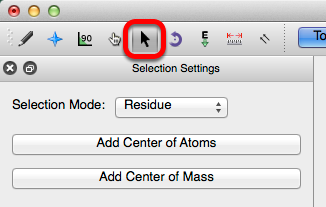
Hay tres tipos de modos de selección: "Atom / Bond", "Residue" y "Molecule".
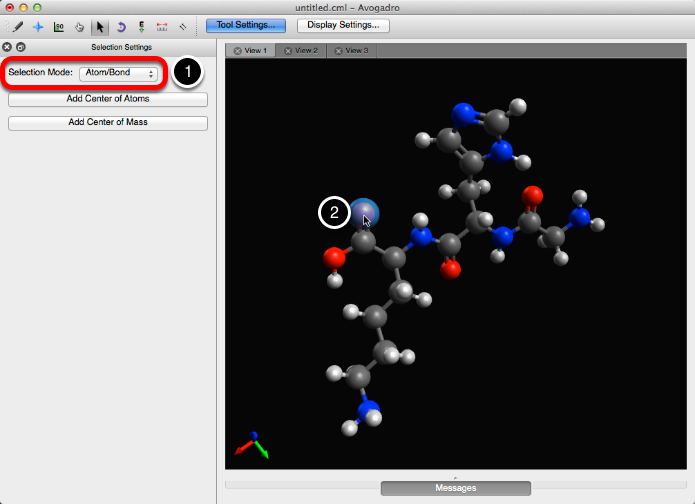
El modo de selección "Atom / Bond" le brinda la posibilidad de seleccionar un solo átomo dentro de una molécula. Esto se logra haciendo clic izquierdo en el átomo. Presionando el botón "Shift" en su teclado permite la selección de múltiples átomos.
Al hacer clic con el botón derecho en la pantalla negra se borrará la selección realizada.
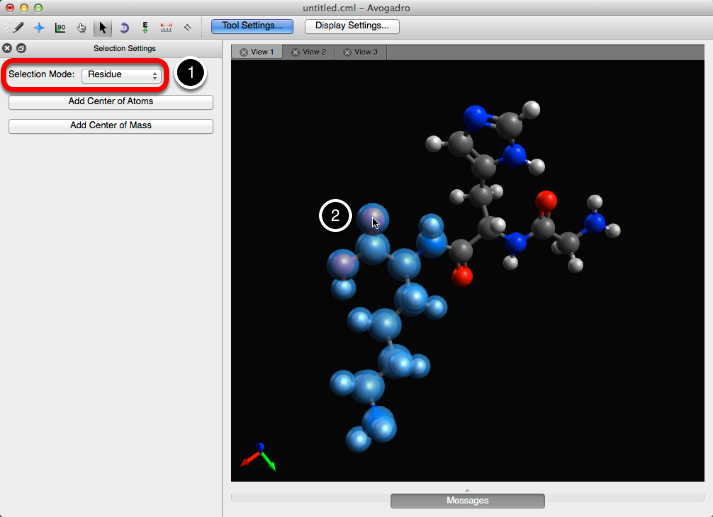
El modo de selección "Residuo" seleccionará un residuo completo dentro de la molécula, haciendo clic en un solo átomo en el residuo.
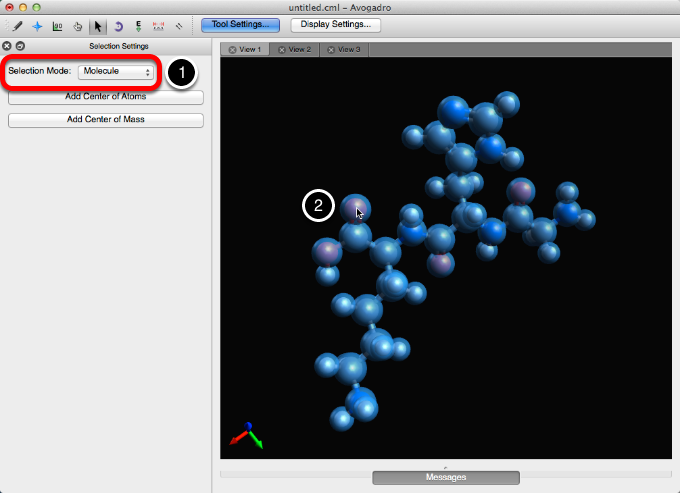
El modo de selección "molécula" seleccionará la molécula completa haciendo clic en un átomo. Hacer doble clic en la molécula también seleccionará la molécula completa.
Las moléculas también se pueden seleccionar haciendo clic y arrastrando el cursor por la pantalla.
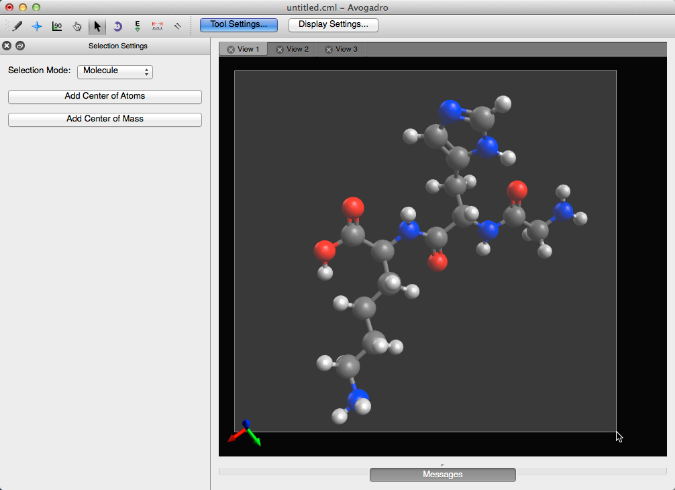
Al hacer clic en "Agregar centro de átomos" se mostrará una bola negra donde se encuentra el centro de la molécula.
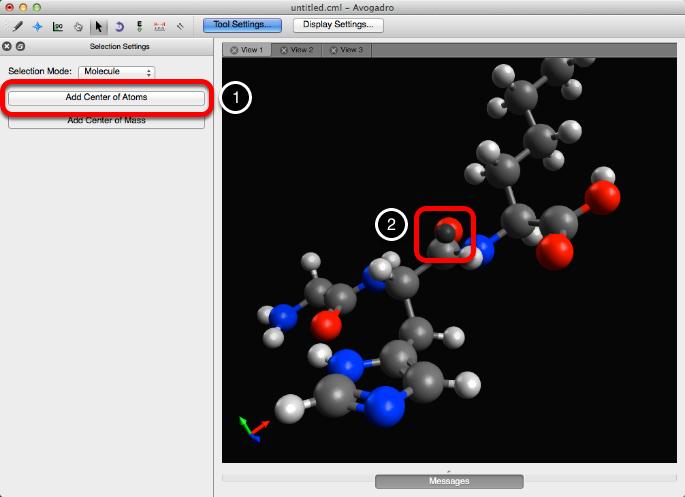
Al hacer clic en "Agregar centro de masa" se mostrará una bola negra donde se encuentra el centro de masa de la molécula.
Herramienta de rotación automática
La herramienta de rotación automática permite la rotación continua de una molécula.
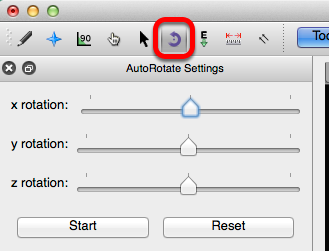
Para rotar la molécula, ajuste las escalas de rotación x, y y z como desee. A continuación, seleccione "Inicio".
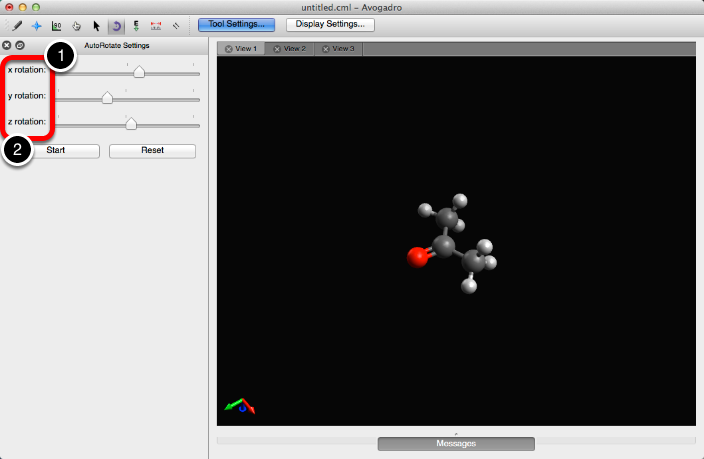
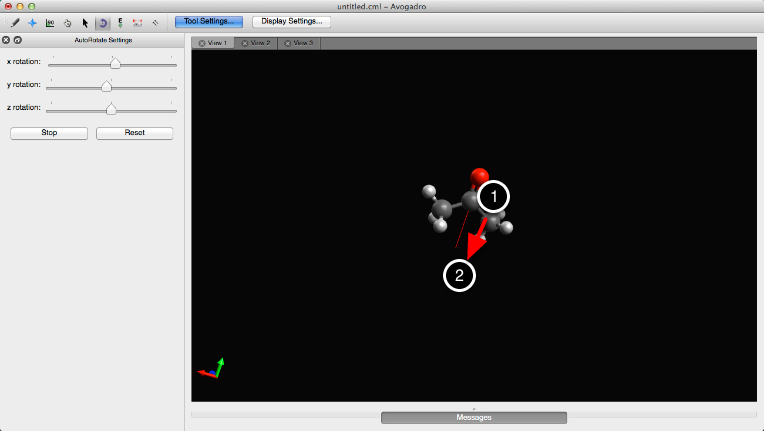
También puede hacer clic y arrastrar desde un átomo para rotar la molécula en una dirección específica.
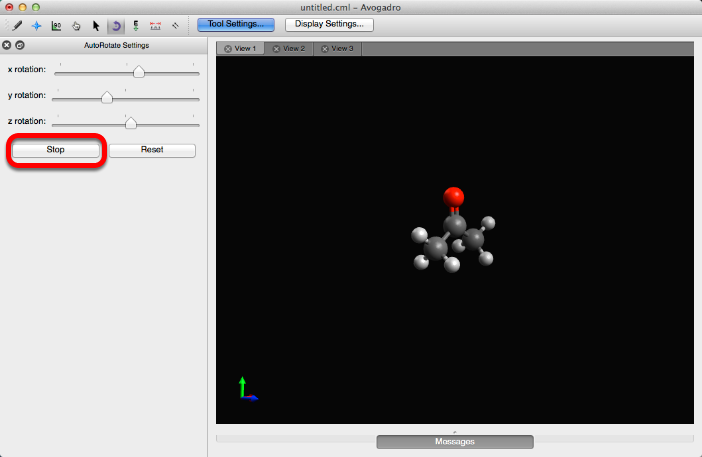
Seleccione "Detener" para terminar la rotación de la molécula.
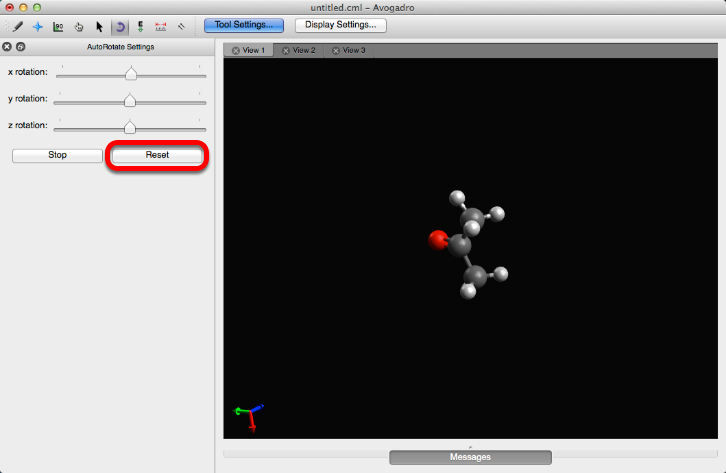
Al hacer clic en "Restablecer" se ajustarán a cero las escalas de rotación y se detendrá la rotación.
Herramienta de optimización automática
La herramienta Auto Optimize optimiza continuamente la geometría molecular a través de la mecánica molecular. Esta herramienta proporciona una interfaz interactiva, que le permite manipular una molécula mientras se optimiza su geometría molecular.
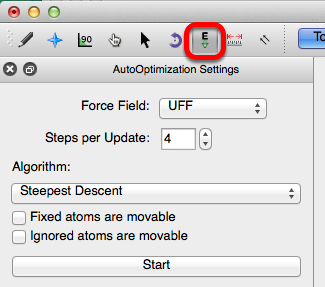
Campos de fuerza y algoritmos
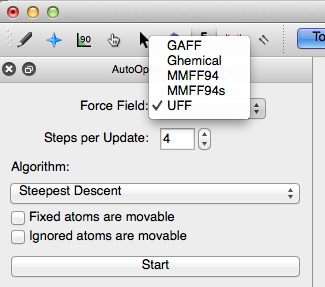
La configuración de optimización automática proporciona varias opciones de campo de fuerza. El campo de fuerza predeterminado en Avogadro es UFF (Campo de fuerza universal). La UFF generalmente puede reproducir la mayoría de las características estructurales en la tabla periódica. Sin embargo, dependiendo de la molécula que se optimice, los otros campos de fuerza pueden ser más adecuados para optimizar los parámetros moleculares. Las opciones de campo de fuerza se muestran a continuación. Para obtener más información sobre los campos de fuerza, consulte la sección de optimización de geometría de este manual de laboratorio.
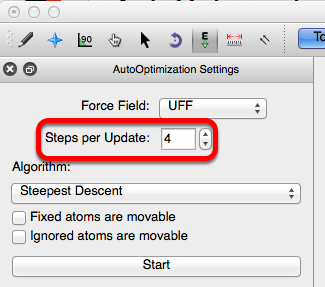
La configuración predeterminada para "Pasos por actualización" es 4. Este número se puede aumentar o disminuir preferentemente. Si tienes una computadora más lenta considera disminuir este número.
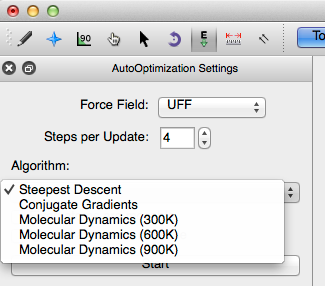
Avogadro también tiene la capacidad de aplicar algoritmos específicos dependiendo de su necesidad. El descenso más pronunciado es el algoritmo predeterminado y tiene el sistema más fluido e interactivo.
Arreglando e ignorando selecciones
Si es necesario, los átomos pueden fijarse en su lugar antes de la optimización para que no se muevan. Para ello, vaya al menú "Extensiones", mantenga el cursor sobre "Mecánica Molecular" y luego seleccione "Arreglar los átomos seleccionados".
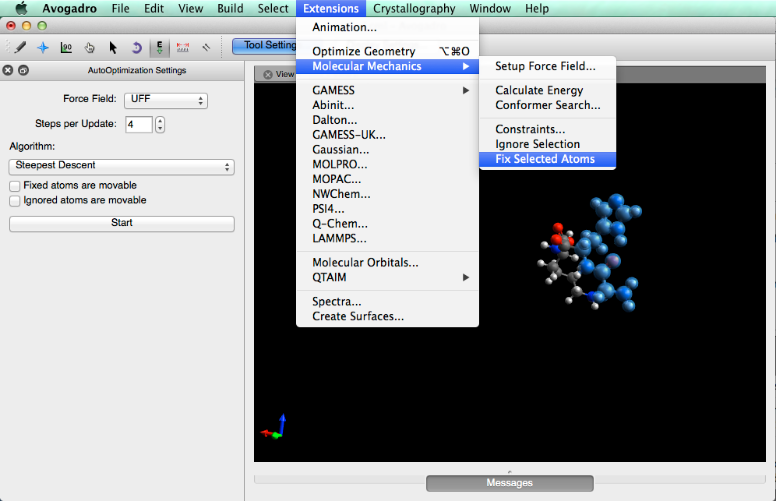
Al hacer clic en "Inicio" le permitirá manipular la molécula haciendo clic con el botón izquierdo en un átomo y arrastrando el cursor. Note que los átomos fijos no se doblan con la manipulación de la molécula.
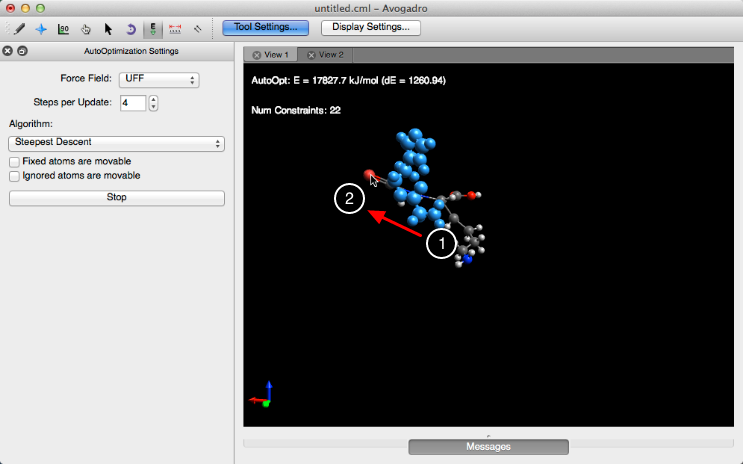
Si la casilla "Los átomos fijos son móviles" está marcada, los enlaces entre los átomos se pueden manipular en una nueva configuración. Manipular un enlace usando esta característica lo bloqueará en su lugar y mostrará el número de restricciones emitidas en esa molécula. A continuación se muestra una versión exagerada de una manipulación de bonos que utiliza esta función.
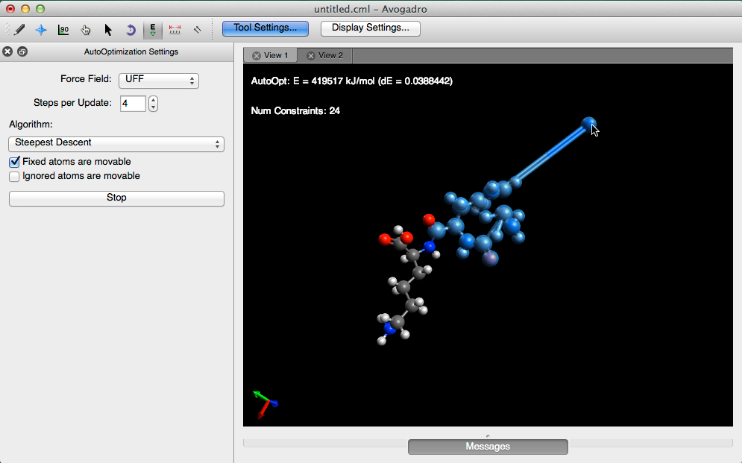
Seleccionando el menú desplegable "Extensiones", manteniendo el cursor sobre la opción "Mecánica Molecular", y seleccionando "Ignorar selección" le permitirá ignorar una selección de átomos. Avogadro registra una selección ignorada como si la selección no existiera. Por lo tanto, la optimización solo tiene lugar en correspondencia con los átomos que no se ignoran. Los átomos que se han ignorado aún se pueden ajustar si la casilla "Los átomos ignorados son móviles" está marcada, sin embargo, no se optimizarán.
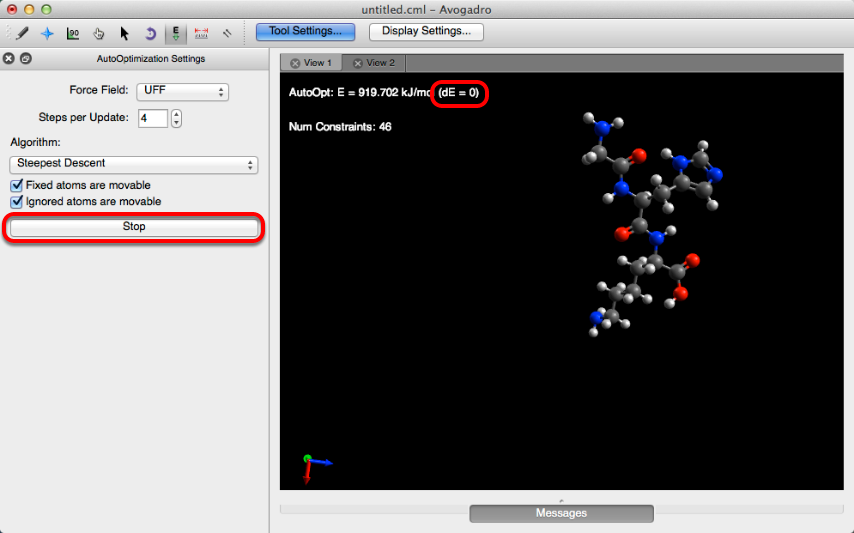
Las moléculas se volverán a optimizar hasta que dE = 0 o se haga clic en "Detener".
Herramienta de medida
La herramienta de medida determina las longitudes de los enlaces, los ángulos y los diédricos.
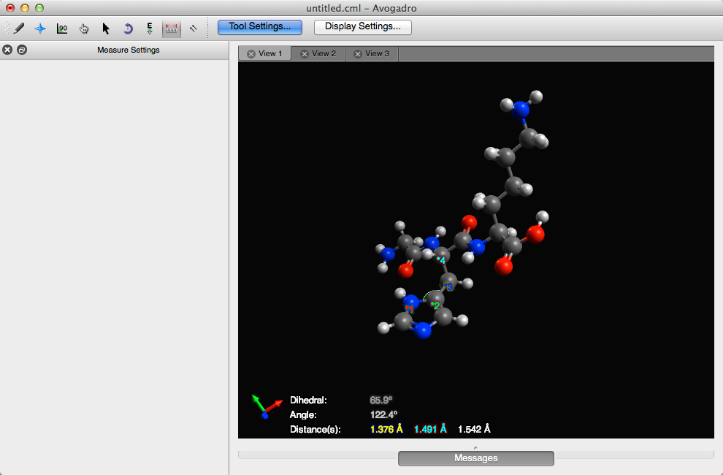
La herramienta de medición le permite seleccionar y evaluar hasta cuatro átomos.
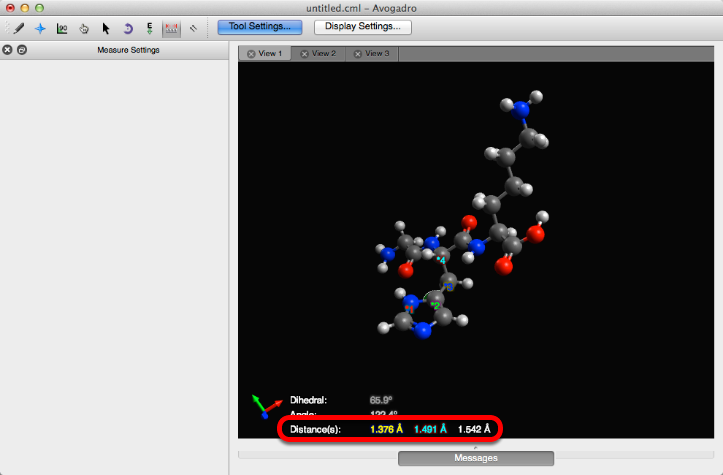
Al hacer clic en los átomos, Avogadro calculará automáticamente las distancias entre los átomos en un orden respectivo. Por ejemplo, la distancia entre el átomo 1 y 2 es 1.376 Å (se muestra a continuación).
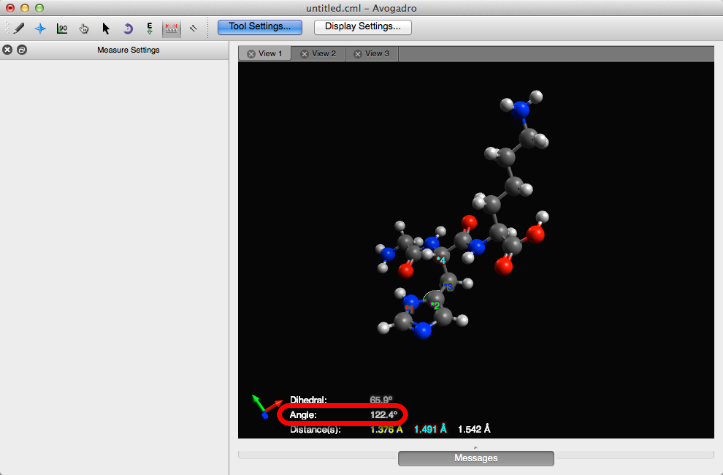
Avogadro también determinará el ángulo entre los átomos, si se han seleccionado al menos tres átomos. El segundo átomo se usa como el vértice.
Si se seleccionan cuatro átomos, se determina un ángulo diedro.
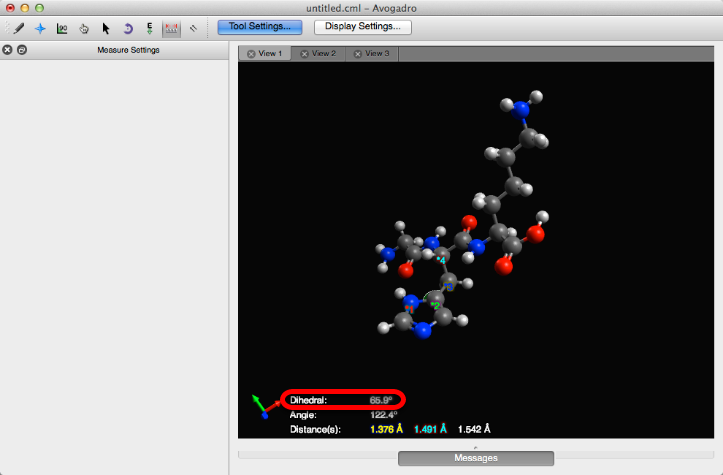
Al hacer clic derecho en la pantalla se restablecerán los átomos previamente seleccionados.
Herramienta de alineación
La herramienta de alineación gira y convierte una (s) molécula (s) en un marco de referencia específico.
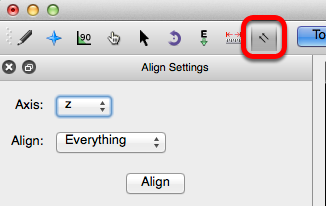
Se puede elegir un eje de alineación en el menú desplegable "Eje". Escribir “x”, “y” o “z” es un atajo para cambiar el eje de alineación.
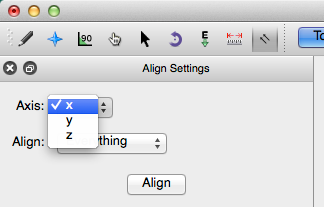
La herramienta de alineación se puede usar para alinear todo en el marco, o una molécula específica con la opción "Molécula" en el menú desplegable "Alinear". Como se muestra a continuación, se utilizarán dos átomos como puntos de referencia para alinear su selección.
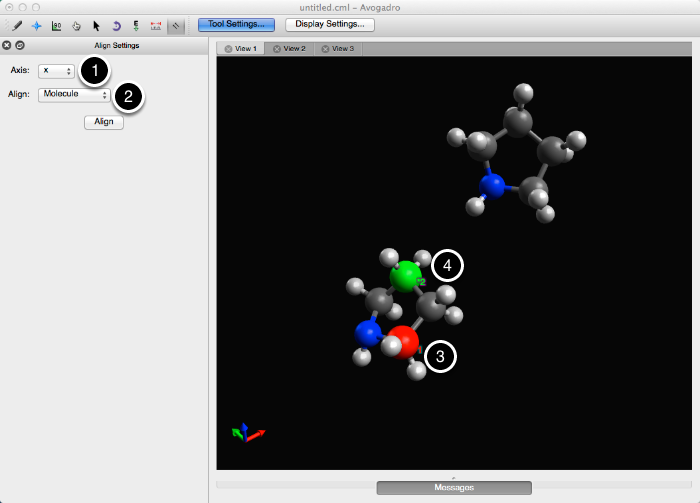
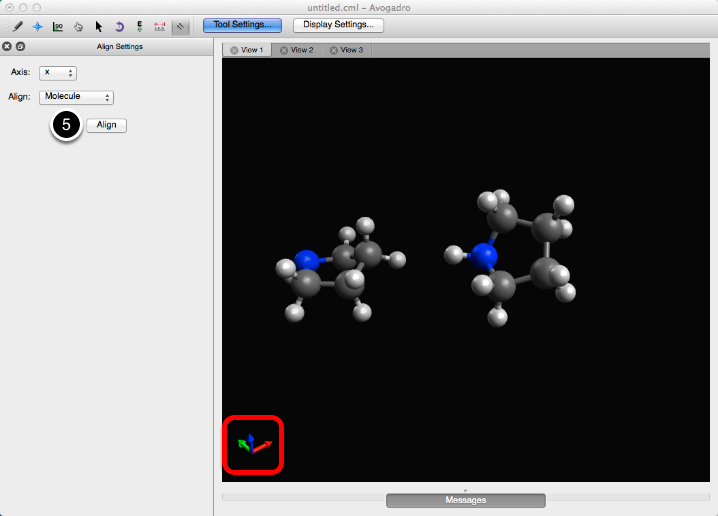
Al hacer clic en "Alinear", se reposicionará la molécula / marco con la nueva alineación. Los ejes de alineación se muestran en la esquina inferior izquierda.
- El eje rojo se designa como el eje X, el verde se designa como el eje Y y el azul se designa como el eje z.
No hay comentarios:
Publicar un comentario